biopipen 0.21.0__py3-none-any.whl → 0.34.26__py3-none-any.whl
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- biopipen/__init__.py +1 -1
- biopipen/core/config.toml +28 -0
- biopipen/core/filters.py +79 -4
- biopipen/core/proc.py +12 -3
- biopipen/core/testing.py +75 -3
- biopipen/ns/bam.py +148 -6
- biopipen/ns/bed.py +75 -0
- biopipen/ns/cellranger.py +186 -0
- biopipen/ns/cellranger_pipeline.py +126 -0
- biopipen/ns/cnv.py +19 -3
- biopipen/ns/cnvkit.py +1 -1
- biopipen/ns/cnvkit_pipeline.py +20 -12
- biopipen/ns/delim.py +34 -35
- biopipen/ns/gene.py +68 -23
- biopipen/ns/gsea.py +63 -37
- biopipen/ns/misc.py +39 -14
- biopipen/ns/plot.py +304 -1
- biopipen/ns/protein.py +183 -0
- biopipen/ns/regulatory.py +290 -0
- biopipen/ns/rnaseq.py +142 -5
- biopipen/ns/scrna.py +2053 -473
- biopipen/ns/scrna_metabolic_landscape.py +228 -382
- biopipen/ns/snp.py +659 -0
- biopipen/ns/stats.py +484 -0
- biopipen/ns/tcr.py +683 -98
- biopipen/ns/vcf.py +236 -2
- biopipen/ns/web.py +97 -6
- biopipen/reports/bam/CNVpytor.svelte +4 -9
- biopipen/reports/cellranger/CellRangerCount.svelte +18 -0
- biopipen/reports/cellranger/CellRangerSummary.svelte +16 -0
- biopipen/reports/cellranger/CellRangerVdj.svelte +18 -0
- biopipen/reports/cnvkit/CNVkitDiagram.svelte +1 -1
- biopipen/reports/cnvkit/CNVkitHeatmap.svelte +1 -1
- biopipen/reports/cnvkit/CNVkitScatter.svelte +1 -1
- biopipen/reports/common.svelte +15 -0
- biopipen/reports/protein/ProdigySummary.svelte +16 -0
- biopipen/reports/scrna/CellsDistribution.svelte +4 -39
- biopipen/reports/scrna/DimPlots.svelte +1 -1
- biopipen/reports/scrna/MarkersFinder.svelte +6 -126
- biopipen/reports/scrna/MetaMarkers.svelte +3 -75
- biopipen/reports/scrna/RadarPlots.svelte +4 -20
- biopipen/reports/scrna_metabolic_landscape/MetabolicFeatures.svelte +61 -22
- biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayActivity.svelte +88 -82
- biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayHeterogeneity.svelte +70 -10
- biopipen/reports/snp/PlinkCallRate.svelte +24 -0
- biopipen/reports/snp/PlinkFreq.svelte +18 -0
- biopipen/reports/snp/PlinkHWE.svelte +18 -0
- biopipen/reports/snp/PlinkHet.svelte +18 -0
- biopipen/reports/snp/PlinkIBD.svelte +18 -0
- biopipen/reports/tcr/CDR3AAPhyschem.svelte +19 -66
- biopipen/reports/tcr/ClonalStats.svelte +16 -0
- biopipen/reports/tcr/CloneResidency.svelte +3 -93
- biopipen/reports/tcr/Immunarch.svelte +4 -155
- biopipen/reports/tcr/TCRClusterStats.svelte +3 -45
- biopipen/reports/tcr/TESSA.svelte +11 -28
- biopipen/reports/utils/misc.liq +22 -7
- biopipen/scripts/bam/BamMerge.py +11 -15
- biopipen/scripts/bam/BamSampling.py +90 -0
- biopipen/scripts/bam/BamSort.py +141 -0
- biopipen/scripts/bam/BamSplitChroms.py +10 -10
- biopipen/scripts/bam/BamSubsetByBed.py +38 -0
- biopipen/scripts/bam/CNAClinic.R +41 -5
- biopipen/scripts/bam/CNVpytor.py +153 -54
- biopipen/scripts/bam/ControlFREEC.py +13 -14
- biopipen/scripts/bam/SamtoolsView.py +33 -0
- biopipen/scripts/bed/Bed2Vcf.py +5 -5
- biopipen/scripts/bed/BedConsensus.py +5 -5
- biopipen/scripts/bed/BedLiftOver.sh +6 -4
- biopipen/scripts/bed/BedtoolsIntersect.py +54 -0
- biopipen/scripts/bed/BedtoolsMakeWindows.py +47 -0
- biopipen/scripts/bed/BedtoolsMerge.py +4 -4
- biopipen/scripts/cellranger/CellRangerCount.py +138 -0
- biopipen/scripts/cellranger/CellRangerSummary.R +181 -0
- biopipen/scripts/cellranger/CellRangerVdj.py +112 -0
- biopipen/scripts/cnv/AneuploidyScore.R +55 -20
- biopipen/scripts/cnv/AneuploidyScoreSummary.R +221 -163
- biopipen/scripts/cnv/TMADScore.R +25 -9
- biopipen/scripts/cnv/TMADScoreSummary.R +57 -86
- biopipen/scripts/cnvkit/CNVkitAccess.py +7 -6
- biopipen/scripts/cnvkit/CNVkitAutobin.py +26 -18
- biopipen/scripts/cnvkit/CNVkitBatch.py +6 -6
- biopipen/scripts/cnvkit/CNVkitCall.py +3 -3
- biopipen/scripts/cnvkit/CNVkitCoverage.py +4 -3
- biopipen/scripts/cnvkit/CNVkitDiagram.py +5 -5
- biopipen/scripts/cnvkit/CNVkitFix.py +3 -3
- biopipen/scripts/cnvkit/CNVkitGuessBaits.py +12 -8
- biopipen/scripts/cnvkit/CNVkitHeatmap.py +5 -5
- biopipen/scripts/cnvkit/CNVkitReference.py +6 -5
- biopipen/scripts/cnvkit/CNVkitScatter.py +5 -5
- biopipen/scripts/cnvkit/CNVkitSegment.py +5 -5
- biopipen/scripts/cnvkit/guess_baits.py +166 -93
- biopipen/scripts/delim/RowsBinder.R +1 -1
- biopipen/scripts/delim/SampleInfo.R +116 -118
- biopipen/scripts/gene/GeneNameConversion.R +67 -0
- biopipen/scripts/gene/GenePromoters.R +61 -0
- biopipen/scripts/gsea/Enrichr.R +5 -5
- biopipen/scripts/gsea/FGSEA.R +184 -50
- biopipen/scripts/gsea/GSEA.R +2 -2
- biopipen/scripts/gsea/PreRank.R +5 -5
- biopipen/scripts/misc/Config2File.py +2 -2
- biopipen/scripts/misc/Plot.R +80 -0
- biopipen/scripts/misc/Shell.sh +15 -0
- biopipen/scripts/misc/Str2File.py +2 -2
- biopipen/scripts/plot/Heatmap.R +3 -3
- biopipen/scripts/plot/Manhattan.R +147 -0
- biopipen/scripts/plot/QQPlot.R +146 -0
- biopipen/scripts/plot/ROC.R +88 -0
- biopipen/scripts/plot/Scatter.R +112 -0
- biopipen/scripts/plot/VennDiagram.R +5 -9
- biopipen/scripts/protein/MMCIF2PDB.py +33 -0
- biopipen/scripts/protein/PDB2Fasta.py +60 -0
- biopipen/scripts/protein/Prodigy.py +119 -0
- biopipen/scripts/protein/ProdigySummary.R +140 -0
- biopipen/scripts/protein/RMSD.py +178 -0
- biopipen/scripts/regulatory/MotifAffinityTest.R +102 -0
- biopipen/scripts/regulatory/MotifAffinityTest_AtSNP.R +127 -0
- biopipen/scripts/regulatory/MotifAffinityTest_MotifBreakR.R +104 -0
- biopipen/scripts/regulatory/MotifScan.py +159 -0
- biopipen/scripts/regulatory/VariantMotifPlot.R +78 -0
- biopipen/scripts/regulatory/motifs-common.R +324 -0
- biopipen/scripts/rnaseq/Simulation-ESCO.R +180 -0
- biopipen/scripts/rnaseq/Simulation-RUVcorr.R +45 -0
- biopipen/scripts/rnaseq/Simulation.R +21 -0
- biopipen/scripts/rnaseq/UnitConversion.R +325 -54
- biopipen/scripts/scrna/AnnData2Seurat.R +40 -0
- biopipen/scripts/scrna/CCPlotR-patch.R +161 -0
- biopipen/scripts/scrna/CellCellCommunication.py +150 -0
- biopipen/scripts/scrna/CellCellCommunicationPlots.R +93 -0
- biopipen/scripts/scrna/CellSNPLite.py +30 -0
- biopipen/scripts/scrna/CellTypeAnnotation-celltypist.R +185 -0
- biopipen/scripts/scrna/CellTypeAnnotation-direct.R +68 -31
- biopipen/scripts/scrna/CellTypeAnnotation-hitype.R +27 -22
- biopipen/scripts/scrna/CellTypeAnnotation-sccatch.R +28 -20
- biopipen/scripts/scrna/CellTypeAnnotation-sctype.R +48 -25
- biopipen/scripts/scrna/CellTypeAnnotation.R +37 -1
- biopipen/scripts/scrna/CellsDistribution.R +456 -167
- biopipen/scripts/scrna/DimPlots.R +1 -1
- biopipen/scripts/scrna/ExprImputation-alra.R +109 -0
- biopipen/scripts/scrna/ExprImputation-rmagic.R +256 -0
- biopipen/scripts/scrna/{ExprImpution-scimpute.R → ExprImputation-scimpute.R} +8 -5
- biopipen/scripts/scrna/ExprImputation.R +7 -0
- biopipen/scripts/scrna/LoomTo10X.R +51 -0
- biopipen/scripts/scrna/MQuad.py +25 -0
- biopipen/scripts/scrna/MarkersFinder.R +679 -400
- biopipen/scripts/scrna/MetaMarkers.R +265 -161
- biopipen/scripts/scrna/ModuleScoreCalculator.R +66 -11
- biopipen/scripts/scrna/PseudoBulkDEG.R +678 -0
- biopipen/scripts/scrna/RadarPlots.R +355 -134
- biopipen/scripts/scrna/ScFGSEA.R +298 -100
- biopipen/scripts/scrna/ScSimulation.R +65 -0
- biopipen/scripts/scrna/ScVelo.py +617 -0
- biopipen/scripts/scrna/Seurat2AnnData.R +7 -0
- biopipen/scripts/scrna/SeuratClusterStats-clustree.R +87 -0
- biopipen/scripts/scrna/SeuratClusterStats-dimplots.R +36 -30
- biopipen/scripts/scrna/SeuratClusterStats-features.R +138 -187
- biopipen/scripts/scrna/SeuratClusterStats-ngenes.R +81 -0
- biopipen/scripts/scrna/SeuratClusterStats-stats.R +78 -89
- biopipen/scripts/scrna/SeuratClusterStats.R +47 -10
- biopipen/scripts/scrna/SeuratClustering.R +36 -233
- biopipen/scripts/scrna/SeuratLoading.R +2 -2
- biopipen/scripts/scrna/SeuratMap2Ref.R +84 -113
- biopipen/scripts/scrna/SeuratMetadataMutater.R +16 -6
- biopipen/scripts/scrna/SeuratPreparing.R +223 -173
- biopipen/scripts/scrna/SeuratSubClustering.R +64 -0
- biopipen/scripts/scrna/SeuratTo10X.R +27 -0
- biopipen/scripts/scrna/Slingshot.R +65 -0
- biopipen/scripts/scrna/Subset10X.R +2 -2
- biopipen/scripts/scrna/TopExpressingGenes.R +169 -135
- biopipen/scripts/scrna/celltypist-wrapper.py +195 -0
- biopipen/scripts/scrna/scvelo_paga.py +313 -0
- biopipen/scripts/scrna/seurat_anndata_conversion.py +98 -0
- biopipen/scripts/scrna_metabolic_landscape/MetabolicFeatures.R +447 -82
- biopipen/scripts/scrna_metabolic_landscape/MetabolicPathwayActivity.R +348 -241
- biopipen/scripts/scrna_metabolic_landscape/MetabolicPathwayHeterogeneity.R +188 -166
- biopipen/scripts/snp/MatrixEQTL.R +217 -0
- biopipen/scripts/snp/Plink2GTMat.py +148 -0
- biopipen/scripts/snp/PlinkCallRate.R +199 -0
- biopipen/scripts/snp/PlinkFilter.py +100 -0
- biopipen/scripts/snp/PlinkFreq.R +291 -0
- biopipen/scripts/snp/PlinkFromVcf.py +81 -0
- biopipen/scripts/snp/PlinkHWE.R +85 -0
- biopipen/scripts/snp/PlinkHet.R +96 -0
- biopipen/scripts/snp/PlinkIBD.R +196 -0
- biopipen/scripts/snp/PlinkSimulation.py +124 -0
- biopipen/scripts/snp/PlinkUpdateName.py +124 -0
- biopipen/scripts/stats/ChowTest.R +146 -0
- biopipen/scripts/stats/DiffCoexpr.R +152 -0
- biopipen/scripts/stats/LiquidAssoc.R +135 -0
- biopipen/scripts/stats/Mediation.R +108 -0
- biopipen/scripts/stats/MetaPvalue.R +130 -0
- biopipen/scripts/stats/MetaPvalue1.R +74 -0
- biopipen/scripts/tcgamaf/Maf2Vcf.py +2 -2
- biopipen/scripts/tcgamaf/MafAddChr.py +2 -2
- biopipen/scripts/tcr/Attach2Seurat.R +3 -2
- biopipen/scripts/tcr/CDR3AAPhyschem.R +211 -143
- biopipen/scripts/tcr/CDR3Clustering.R +343 -0
- biopipen/scripts/tcr/ClonalStats.R +526 -0
- biopipen/scripts/tcr/CloneResidency.R +255 -131
- biopipen/scripts/tcr/CloneSizeQQPlot.R +4 -4
- biopipen/scripts/tcr/GIANA/GIANA.py +1356 -797
- biopipen/scripts/tcr/GIANA/GIANA4.py +1362 -789
- biopipen/scripts/tcr/GIANA/query.py +164 -162
- biopipen/scripts/tcr/Immunarch-basic.R +31 -9
- biopipen/scripts/tcr/Immunarch-clonality.R +25 -5
- biopipen/scripts/tcr/Immunarch-diversity.R +352 -134
- biopipen/scripts/tcr/Immunarch-geneusage.R +45 -5
- biopipen/scripts/tcr/Immunarch-kmer.R +68 -8
- biopipen/scripts/tcr/Immunarch-overlap.R +84 -4
- biopipen/scripts/tcr/Immunarch-spectratyping.R +35 -6
- biopipen/scripts/tcr/Immunarch-tracking.R +38 -6
- biopipen/scripts/tcr/Immunarch-vjjunc.R +165 -0
- biopipen/scripts/tcr/Immunarch.R +63 -11
- biopipen/scripts/tcr/Immunarch2VDJtools.R +2 -2
- biopipen/scripts/tcr/ImmunarchFilter.R +4 -4
- biopipen/scripts/tcr/ImmunarchLoading.R +38 -29
- biopipen/scripts/tcr/SampleDiversity.R +1 -1
- biopipen/scripts/tcr/ScRepCombiningExpression.R +40 -0
- biopipen/scripts/tcr/ScRepLoading.R +166 -0
- biopipen/scripts/tcr/TCRClusterStats.R +176 -22
- biopipen/scripts/tcr/TCRDock.py +110 -0
- biopipen/scripts/tcr/TESSA.R +102 -118
- biopipen/scripts/tcr/VJUsage.R +5 -5
- biopipen/scripts/tcr/immunarch-patched.R +142 -0
- biopipen/scripts/tcr/vdjtools-patch.sh +1 -1
- biopipen/scripts/vcf/BcftoolsAnnotate.py +91 -0
- biopipen/scripts/vcf/BcftoolsFilter.py +90 -0
- biopipen/scripts/vcf/BcftoolsMerge.py +31 -0
- biopipen/scripts/vcf/BcftoolsSort.py +113 -0
- biopipen/scripts/vcf/BcftoolsView.py +73 -0
- biopipen/scripts/vcf/TruvariBench.sh +14 -7
- biopipen/scripts/vcf/TruvariBenchSummary.R +16 -13
- biopipen/scripts/vcf/TruvariConsistency.R +1 -1
- biopipen/scripts/vcf/Vcf2Bed.py +2 -2
- biopipen/scripts/vcf/VcfAnno.py +11 -11
- biopipen/scripts/vcf/VcfDownSample.sh +22 -10
- biopipen/scripts/vcf/VcfFilter.py +5 -5
- biopipen/scripts/vcf/VcfFix.py +7 -7
- biopipen/scripts/vcf/VcfFix_utils.py +13 -4
- biopipen/scripts/vcf/VcfIndex.py +3 -3
- biopipen/scripts/vcf/VcfIntersect.py +3 -3
- biopipen/scripts/vcf/VcfLiftOver.sh +5 -0
- biopipen/scripts/vcf/VcfSplitSamples.py +4 -4
- biopipen/scripts/vcf/bcftools_utils.py +52 -0
- biopipen/scripts/web/Download.py +8 -4
- biopipen/scripts/web/DownloadList.py +5 -5
- biopipen/scripts/web/GCloudStorageDownloadBucket.py +82 -0
- biopipen/scripts/web/GCloudStorageDownloadFile.py +23 -0
- biopipen/scripts/web/gcloud_common.py +49 -0
- biopipen/utils/gene.py +108 -60
- biopipen/utils/misc.py +146 -20
- biopipen/utils/reference.py +64 -20
- biopipen/utils/reporter.py +177 -0
- biopipen/utils/vcf.py +1 -1
- biopipen-0.34.26.dist-info/METADATA +27 -0
- biopipen-0.34.26.dist-info/RECORD +292 -0
- {biopipen-0.21.0.dist-info → biopipen-0.34.26.dist-info}/WHEEL +1 -1
- {biopipen-0.21.0.dist-info → biopipen-0.34.26.dist-info}/entry_points.txt +6 -2
- biopipen/ns/bcftools.py +0 -111
- biopipen/ns/scrna_basic.py +0 -255
- biopipen/reports/delim/SampleInfo.svelte +0 -36
- biopipen/reports/scrna/GeneExpressionInvistigation.svelte +0 -32
- biopipen/reports/scrna/ScFGSEA.svelte +0 -35
- biopipen/reports/scrna/SeuratClusterStats.svelte +0 -82
- biopipen/reports/scrna/SeuratMap2Ref.svelte +0 -20
- biopipen/reports/scrna/SeuratPreparing.svelte +0 -38
- biopipen/reports/scrna/TopExpressingGenes.svelte +0 -55
- biopipen/reports/scrna_metabolic_landscape/MetabolicFeaturesIntraSubset.svelte +0 -31
- biopipen/reports/utils/gsea.liq +0 -110
- biopipen/scripts/bcftools/BcftoolsAnnotate.py +0 -42
- biopipen/scripts/bcftools/BcftoolsFilter.py +0 -79
- biopipen/scripts/bcftools/BcftoolsSort.py +0 -19
- biopipen/scripts/gene/GeneNameConversion.py +0 -66
- biopipen/scripts/scrna/ExprImpution-alra.R +0 -32
- biopipen/scripts/scrna/ExprImpution-rmagic.R +0 -29
- biopipen/scripts/scrna/ExprImpution.R +0 -7
- biopipen/scripts/scrna/GeneExpressionInvistigation.R +0 -132
- biopipen/scripts/scrna/Write10X.R +0 -11
- biopipen/scripts/scrna_metabolic_landscape/MetabolicFeaturesIntraSubset.R +0 -150
- biopipen/scripts/tcr/TCRClustering.R +0 -280
- biopipen/utils/common_docstrs.py +0 -61
- biopipen/utils/gene.R +0 -49
- biopipen/utils/gsea.R +0 -193
- biopipen/utils/io.R +0 -20
- biopipen/utils/misc.R +0 -114
- biopipen/utils/mutate_helpers.R +0 -433
- biopipen/utils/plot.R +0 -173
- biopipen/utils/rnaseq.R +0 -48
- biopipen/utils/single_cell.R +0 -115
- biopipen-0.21.0.dist-info/METADATA +0 -22
- biopipen-0.21.0.dist-info/RECORD +0 -218
biopipen/ns/tcr.py
CHANGED
|
@@ -1,10 +1,11 @@
|
|
|
1
1
|
"""Tools to analyze single-cell TCR sequencing data"""
|
|
2
|
-
|
|
2
|
+
from pipen.utils import mark
|
|
3
3
|
from ..core.defaults import SCRIPT_DIR
|
|
4
4
|
from ..core.proc import Proc
|
|
5
5
|
from ..core.config import config
|
|
6
6
|
|
|
7
7
|
|
|
8
|
+
@mark(deprecated="{proc.name} is deprecated, use ScRepLoading instead.")
|
|
8
9
|
class ImmunarchLoading(Proc):
|
|
9
10
|
"""Immuarch - Loading data
|
|
10
11
|
|
|
@@ -39,12 +40,15 @@ class ImmunarchLoading(Proc):
|
|
|
39
40
|
information.
|
|
40
41
|
|
|
41
42
|
Output:
|
|
42
|
-
rdsfile: The RDS file with the data and metadata
|
|
43
|
-
|
|
43
|
+
rdsfile: The RDS file with the data and metadata, which can be processed by
|
|
44
|
+
other `immunarch` functions.
|
|
45
|
+
metatxt: The meta data at cell level, which can be used to attach to the Seurat object
|
|
44
46
|
|
|
45
47
|
Envs:
|
|
46
48
|
prefix: The prefix to the barcodes. You can use placeholder like `{Sample}_`
|
|
47
|
-
to use the meta data from the `immunarch` object.
|
|
49
|
+
to use the meta data from the `immunarch` object. The prefixed barcodes will
|
|
50
|
+
be saved in `out.metatxt`. The `immunarch` object keeps the original barcodes, but
|
|
51
|
+
the prefix is saved at `immdata$prefix`.
|
|
48
52
|
|
|
49
53
|
/// Note
|
|
50
54
|
This option is useful because the barcodes for the cells from scRNA-seq
|
|
@@ -61,14 +65,20 @@ class ImmunarchLoading(Proc):
|
|
|
61
65
|
are not in the same directory, we can link them to a temporary directory
|
|
62
66
|
and pass the temporary directory to `Immunarch`.
|
|
63
67
|
This option is useful when the data files are in different directories.
|
|
64
|
-
mode
|
|
68
|
+
mode: Either "single" for single chain data or "paired" for
|
|
65
69
|
paired chain data. For `single`, only TRB chain will be kept
|
|
66
70
|
at `immdata$data`, information for other chains will be
|
|
67
71
|
saved at `immdata$tra` and `immdata$multi`.
|
|
68
|
-
|
|
72
|
+
extracols (list): The extra columns to be exported to the text file.
|
|
69
73
|
You can refer to the
|
|
70
74
|
[immunarch documentation](https://immunarch.com/articles/v2_data.html#immunarch-data-format)
|
|
71
|
-
for the full list of the columns.
|
|
75
|
+
to get a sense for the full list of the columns.
|
|
76
|
+
The columns may vary depending on the data source.
|
|
77
|
+
The columns from `immdata$meta` and some core columns, including
|
|
78
|
+
`Barcode`, `CDR3.aa`, `Clones`, `Proportion`, `V.name`, `J.name`, and
|
|
79
|
+
`D.name` will be exported by default. You can use this option to
|
|
80
|
+
specify the extra columns to be exported.
|
|
81
|
+
|
|
72
82
|
""" # noqa: E501
|
|
73
83
|
input = "metafile:file"
|
|
74
84
|
output = [
|
|
@@ -79,12 +89,13 @@ class ImmunarchLoading(Proc):
|
|
|
79
89
|
envs = {
|
|
80
90
|
"tmpdir": config.path.tmpdir,
|
|
81
91
|
"prefix": "{Sample}_",
|
|
82
|
-
"mode": "
|
|
83
|
-
"
|
|
92
|
+
"mode": "paired",
|
|
93
|
+
"extracols": [],
|
|
84
94
|
}
|
|
85
95
|
script = "file://../scripts/tcr/ImmunarchLoading.R"
|
|
86
96
|
|
|
87
97
|
|
|
98
|
+
@mark(deprecated=True)
|
|
88
99
|
class ImmunarchFilter(Proc):
|
|
89
100
|
"""Immunarch - Filter data
|
|
90
101
|
|
|
@@ -163,12 +174,13 @@ class ImmunarchFilter(Proc):
|
|
|
163
174
|
script = "file://../scripts/tcr/ImmunarchFilter.R"
|
|
164
175
|
|
|
165
176
|
|
|
177
|
+
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
166
178
|
class Immunarch(Proc):
|
|
167
179
|
"""Exploration of Single-cell and Bulk T-cell/Antibody Immune Repertoires
|
|
168
180
|
|
|
169
181
|
See <https://immunarch.com/articles/web_only/v3_basic_analysis.html>
|
|
170
182
|
|
|
171
|
-
After [`ImmunarchLoading`](
|
|
183
|
+
After [`ImmunarchLoading`](!!#biopipennstcrimmunarchloading) loads the raw data into an [immunarch](https://immunarch.com) object,
|
|
172
184
|
this process wraps the functions from [`immunarch`](https://immunarch.com) to do the following:
|
|
173
185
|
|
|
174
186
|
- Basic statistics, provided by [`immunarch::repExplore`](https://immunarch.com/reference/repExplore.html), such as number of clones or distributions of lengths and counts.
|
|
@@ -180,6 +192,7 @@ class Immunarch(Proc):
|
|
|
180
192
|
- The dynamics of repertoires across time points/samples, provided by [`immunarch::trackClonotypes`](https://immunarch.com/reference/trackClonotypes.html)
|
|
181
193
|
- The spectratype of clonotypes, provided by [`immunarch::spectratype`](https://immunarch.com/reference/spectratype.html)
|
|
182
194
|
- The distributions of kmers and sequence profiles, provided by [`immunarch::getKmers`](https://immunarch.com/reference/getKmers.html)
|
|
195
|
+
- The V-J junction circos plots, implemented within the script of this process.
|
|
183
196
|
|
|
184
197
|
Environment Variable Design:
|
|
185
198
|
With different sets of arguments, a single function of the above can perform different tasks.
|
|
@@ -218,7 +231,7 @@ class Immunarch(Proc):
|
|
|
218
231
|
vis_args = { "-plot": "heatmap2" }
|
|
219
232
|
```
|
|
220
233
|
|
|
221
|
-
`-plot` will be translated to `.plot` and then passed to `vis`.
|
|
234
|
+
`-plot` will be translated to `.plot` and then passed to `vis`.
|
|
222
235
|
|
|
223
236
|
If multiple cases share the same arguments, we can use the following configuration:
|
|
224
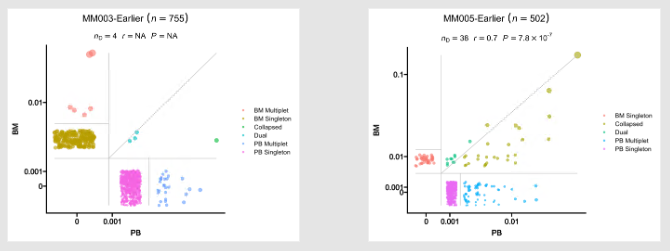
237
|
|
|
@@ -321,6 +334,7 @@ class Immunarch(Proc):
|
|
|
321
334
|
prefix: The prefix to the barcodes. You can use placeholder like `{Sample}_`
|
|
322
335
|
The prefixed barcodes will be used to match the barcodes in `in.metafile`.
|
|
323
336
|
Not used if `in.metafile` is not specified.
|
|
337
|
+
If `None` (default), `immdata$prefix` will be used.
|
|
324
338
|
volumes (ns): Explore clonotype volume (sizes).
|
|
325
339
|
- by: Groupings when visualize clonotype volumes, passed to the `.by` argument of `vis(imm_vol, .by = <values>)`.
|
|
326
340
|
Multiple columns should be separated by `,`.
|
|
@@ -553,12 +567,13 @@ class Immunarch(Proc):
|
|
|
553
567
|
A Gini coefficient of one (or 100 percents) expresses maximal inequality among values (for example where only one person has all the income).
|
|
554
568
|
- d50: The D50 index.
|
|
555
569
|
It is the number of types that are needed to cover 50%% of the total abundance.
|
|
556
|
-
- dxx: The Dxx index.
|
|
557
|
-
It is the number of types that are needed to cover xx%% of the total abundance.
|
|
558
|
-
The percentage should be specified in the `args` argument using `perc` key.
|
|
559
570
|
- raref: Species richness from the results of sampling through extrapolation.
|
|
560
571
|
- by: The variables (column names) to group samples.
|
|
561
572
|
Multiple columns should be separated by `,`.
|
|
573
|
+
- plot_type (choice): The type of the plot, works when `by` is specified.
|
|
574
|
+
Not working for `raref`.
|
|
575
|
+
- box: Boxplot
|
|
576
|
+
- bar: Barplot with error bars
|
|
562
577
|
- subset: Subset the data before calculating the clonotype volumes.
|
|
563
578
|
The whole data will be expanded to cell level, and then subsetted.
|
|
564
579
|
Clone sizes will be re-calculated based on the subsetted data.
|
|
@@ -586,6 +601,13 @@ class Immunarch(Proc):
|
|
|
586
601
|
- fdr: Benjamini & Hochberg (non-negative)
|
|
587
602
|
- none: no correction.
|
|
588
603
|
- separate_by: A column name used to separate the samples into different plots.
|
|
604
|
+
- split_by: A column name used to split the samples into different subplots.
|
|
605
|
+
Like `separate_by`, but the plots will be put in the same figure.
|
|
606
|
+
y-axis will be shared, even if `align_y` is `False` or `ymin`/`ymax` are not specified.
|
|
607
|
+
`ncol` will be ignored.
|
|
608
|
+
- split_order: The order of the values in `split_by` on the x-axis of the plots.
|
|
609
|
+
It can also be used for `separate_by` to control the order of the plots.
|
|
610
|
+
Values can be separated by `,`.
|
|
589
611
|
- align_x (flag): Align the x-axis of multiple plots. Only works for `raref`.
|
|
590
612
|
- align_y (flag): Align the y-axis of multiple plots.
|
|
591
613
|
- ymin (type=float): The minimum value of the y-axis.
|
|
@@ -657,13 +679,31 @@ class Immunarch(Proc):
|
|
|
657
679
|
The values will be passed to the corresponding arguments above.
|
|
658
680
|
If any of these arguments are not specified, the default case will be added, with the name `DEFAULT` and the
|
|
659
681
|
values of `envs.kmers.k`, `envs.kmers.head`, `envs.kmers.vis_args` and `envs.kmers.devpars`.
|
|
682
|
+
vj_junc (ns): Arguments for VJ junction circos plots.
|
|
683
|
+
This analysis is not included in `immunarch`. It is a separate implementation using [`circlize`](https://github.com/jokergoo/circlize).
|
|
684
|
+
- by: Groupings to show VJ usages. Typically, this is the `Sample` column, so that the VJ usages are shown for each sample.
|
|
685
|
+
But you can also use other columns, such as `Subject` to show the VJ usages for each subject.
|
|
686
|
+
Multiple columns should be separated by `,`.
|
|
687
|
+
- by_clones (flag): If True, the VJ usages will be calculated based on the distinct clonotypes, instead of the individual cells.
|
|
688
|
+
- subset: Subset the data before plotting VJ usages.
|
|
689
|
+
The whole data will be expanded to cell level, and then subsetted.
|
|
690
|
+
Clone sizes will be re-calculated based on the subsetted data, which will affect the VJ usages at cell level (by_clones=False).
|
|
691
|
+
- devpars (ns): The parameters for the plotting device.
|
|
692
|
+
- width (type=int): The width of the plot.
|
|
693
|
+
- height (type=int): The height of the plot.
|
|
694
|
+
- res (type=int): The resolution of the plot.
|
|
695
|
+
- cases (type=json;order=9): If you have multiple cases, you can use this argument to specify them.
|
|
696
|
+
The keys will be used as the names of the cases. The values will be passed to the corresponding arguments above.
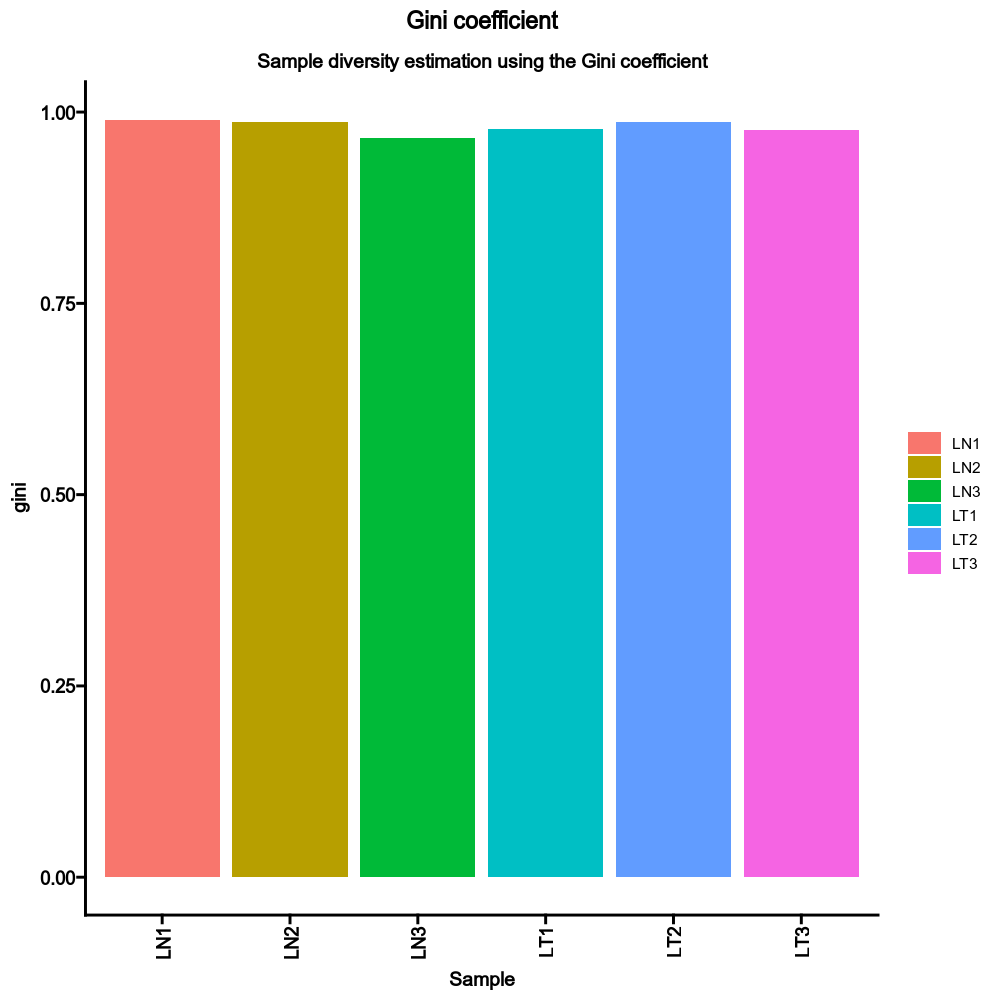
|
|
697
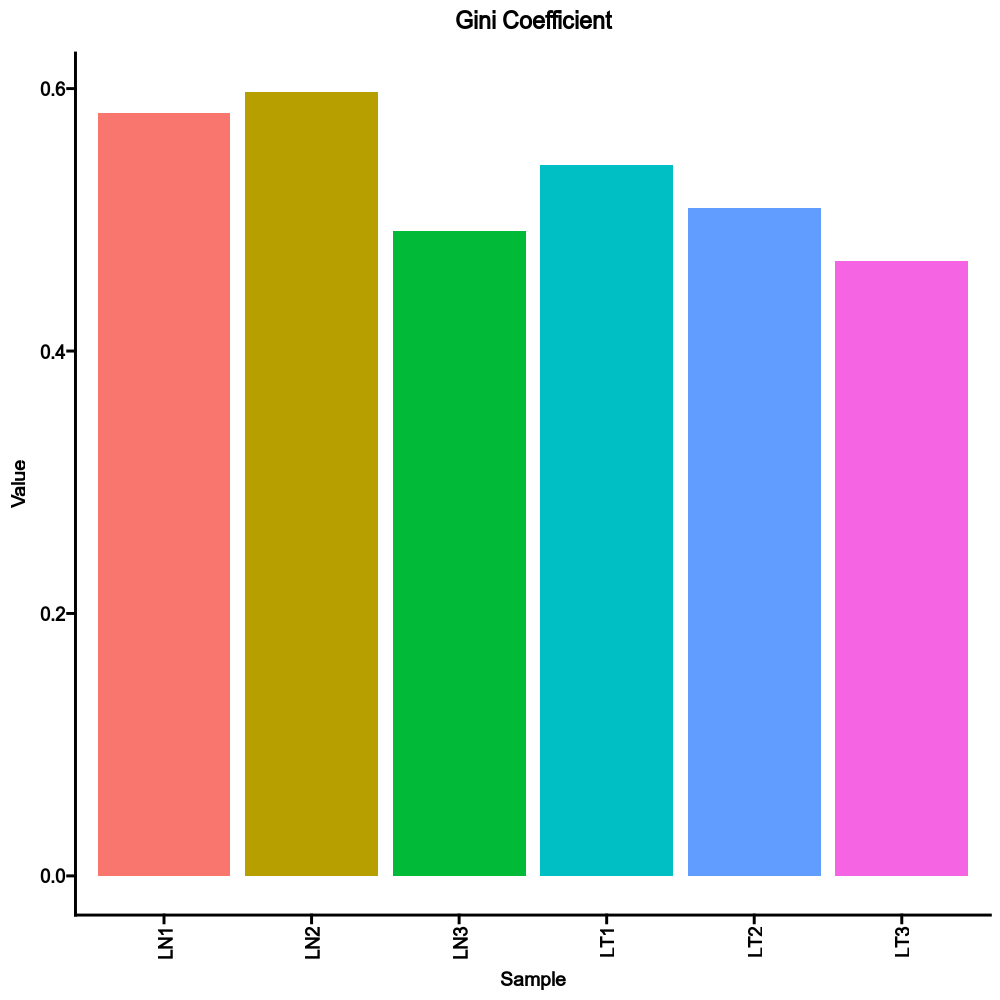
|
+
If any of these arguments are not specified, the values in `envs.vj_junc` will be used.
|
|
698
|
+
If NO cases are specified, the default case will be added, with the name `DEFAULT` and the
|
|
699
|
+
values of `envs.vj_junc.by`, `envs.vj_junc.by_clones` `envs.vj_junc.subset` and `envs.vj_junc.devpars`.
|
|
660
700
|
""" # noqa: E501
|
|
661
701
|
input = "immdata:file,metafile:file"
|
|
662
702
|
output = "outdir:dir:{{in.immdata | stem}}.immunarch"
|
|
663
703
|
lang = config.lang.rscript
|
|
664
704
|
envs = {
|
|
665
705
|
"mutaters": {},
|
|
666
|
-
"prefix":
|
|
706
|
+
"prefix": None,
|
|
667
707
|
# basic statistics
|
|
668
708
|
"volumes": {
|
|
669
709
|
"by": None,
|
|
@@ -754,9 +794,9 @@ class Immunarch(Proc):
|
|
|
754
794
|
},
|
|
755
795
|
# Diversity
|
|
756
796
|
"divs": {
|
|
757
|
-
"filter": None,
|
|
758
797
|
"method": "gini",
|
|
759
798
|
"by": None,
|
|
799
|
+
"plot_type": "bar",
|
|
760
800
|
"args": {},
|
|
761
801
|
"order": [],
|
|
762
802
|
"test": {
|
|
@@ -764,12 +804,14 @@ class Immunarch(Proc):
|
|
|
764
804
|
"padjust": "none",
|
|
765
805
|
},
|
|
766
806
|
"separate_by": None,
|
|
807
|
+
"split_by": None,
|
|
808
|
+
"split_order": None,
|
|
767
809
|
"align_x": False,
|
|
768
810
|
"align_y": False,
|
|
769
811
|
"log": False,
|
|
770
812
|
"devpars": {
|
|
771
|
-
"width":
|
|
772
|
-
"height":
|
|
813
|
+
"width": 800,
|
|
814
|
+
"height": 800,
|
|
773
815
|
"res": 100,
|
|
774
816
|
},
|
|
775
817
|
"subset": None,
|
|
@@ -801,14 +843,24 @@ class Immunarch(Proc):
|
|
|
801
843
|
},
|
|
802
844
|
"cases": {},
|
|
803
845
|
},
|
|
846
|
+
# VJ junction
|
|
847
|
+
"vj_junc": {
|
|
848
|
+
"by": "Sample",
|
|
849
|
+
"by_clones": True,
|
|
850
|
+
"devpars": {"width": 800, "height": 800, "res": 100},
|
|
851
|
+
"subset": None,
|
|
852
|
+
"cases": {},
|
|
853
|
+
},
|
|
804
854
|
}
|
|
805
855
|
script = "file://../scripts/tcr/Immunarch.R"
|
|
806
856
|
plugin_opts = {
|
|
807
857
|
"report": "file://../reports/tcr/Immunarch.svelte",
|
|
808
858
|
"report_paging": 3,
|
|
859
|
+
"poplog_max": 999,
|
|
809
860
|
}
|
|
810
861
|
|
|
811
862
|
|
|
863
|
+
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
812
864
|
class SampleDiversity(Proc):
|
|
813
865
|
"""Sample diversity and rarefaction analysis
|
|
814
866
|
|
|
@@ -857,6 +909,7 @@ class SampleDiversity(Proc):
|
|
|
857
909
|
}
|
|
858
910
|
|
|
859
911
|
|
|
912
|
+
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
860
913
|
class CloneResidency(Proc):
|
|
861
914
|
"""Identification of clone residency
|
|
862
915
|
|
|
@@ -876,7 +929,7 @@ class CloneResidency(Proc):
|
|
|
876
929
|
|
|
877
930
|
- Residency plots showing the residency of clones in the two groups
|
|
878
931
|
|
|
879
|
-

|
|
932
|
+
|
|
880
933
|
|
|
881
934
|
The points in the plot are jittered to avoid overplotting. The x-axis is the residency in the first group and
|
|
882
935
|
the y-axis is the residency in the second group. The size of the points are relative to the normalized size of
|
|
@@ -896,7 +949,7 @@ class CloneResidency(Proc):
|
|
|
896
949
|
|
|
897
950
|
- Venn diagrams showing the overlap of the clones in the two groups
|
|
898
951
|
|
|
899
|
-
{: width="60%"}
|
|
952
|
+
{: width="60%"}
|
|
900
953
|
|
|
901
954
|
Input:
|
|
902
955
|
immdata: The data loaded by `immunarch::repLoad()`
|
|
@@ -936,6 +989,12 @@ class CloneResidency(Proc):
|
|
|
936
989
|
before calculating the clone residency. For example, `Clones > 1` to filter
|
|
937
990
|
out singletons.
|
|
938
991
|
prefix: The prefix of the cell barcodes in the `Seurat` object.
|
|
992
|
+
upset_ymax: The maximum value of the y-axis in the upset bar plots.
|
|
993
|
+
upset_trans: The transformation to apply to the y axis of upset bar plots.
|
|
994
|
+
For example, `log10` or `sqrt`. If not specified, the y axis will be
|
|
995
|
+
plotted as is. Note that the position of the bar plots will be dodged
|
|
996
|
+
instead of stacked when the transformation is applied.
|
|
997
|
+
See also <https://github.com/tidyverse/ggplot2/issues/3671>
|
|
939
998
|
cases (type=json): If you have multiple cases, you can use this argument
|
|
940
999
|
to specify them. The keys will be used as the names of the cases.
|
|
941
1000
|
The values will be passed to the corresponding arguments.
|
|
@@ -955,6 +1014,8 @@ class CloneResidency(Proc):
|
|
|
955
1014
|
"mutaters": {},
|
|
956
1015
|
"subset": None,
|
|
957
1016
|
"prefix": "{Sample}_",
|
|
1017
|
+
"upset_ymax": None,
|
|
1018
|
+
"upset_trans": None,
|
|
958
1019
|
"cases": {},
|
|
959
1020
|
}
|
|
960
1021
|
script = "file://../scripts/tcr/CloneResidency.R"
|
|
@@ -962,12 +1023,14 @@ class CloneResidency(Proc):
|
|
|
962
1023
|
plugin_opts = {"report": "file://../reports/tcr/CloneResidency.svelte"}
|
|
963
1024
|
|
|
964
1025
|
|
|
1026
|
+
@mark(deprecated=True)
|
|
965
1027
|
class Immunarch2VDJtools(Proc):
|
|
966
1028
|
"""Convert immuarch format into VDJtools input formats.
|
|
967
1029
|
|
|
968
1030
|
This process converts the [`immunarch`](https://immunarch.com/) object to the
|
|
969
1031
|
[`VDJtools`](https://vdjtools-doc.readthedocs.io/en/master/) input files,
|
|
970
|
-
in order to perform the VJ gene usage analysis by
|
|
1032
|
+
in order to perform the VJ gene usage analysis by
|
|
1033
|
+
[`VJUsage`](!!#biopipennstcrvjusage) process.
|
|
971
1034
|
|
|
972
1035
|
This process will generally generate a tab-delimited file for each sample,
|
|
973
1036
|
with the following columns.
|
|
@@ -997,6 +1060,7 @@ class Immunarch2VDJtools(Proc):
|
|
|
997
1060
|
script = "file://../scripts/tcr/Immunarch2VDJtools.R"
|
|
998
1061
|
|
|
999
1062
|
|
|
1063
|
+
@mark(deprecated=True)
|
|
1000
1064
|
class ImmunarchSplitIdents(Proc):
|
|
1001
1065
|
"""Split the data into multiple immunarch datasets by Idents from Seurat
|
|
1002
1066
|
|
|
@@ -1030,6 +1094,7 @@ class ImmunarchSplitIdents(Proc):
|
|
|
1030
1094
|
script = "file://../scripts/tcr/ImmunarchSplitIdents.R"
|
|
1031
1095
|
|
|
1032
1096
|
|
|
1097
|
+
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
1033
1098
|
class VJUsage(Proc):
|
|
1034
1099
|
"""Circos-style V-J usage plot displaying the frequency of
|
|
1035
1100
|
various V-J junctions using vdjtools.
|
|
@@ -1072,6 +1137,7 @@ class VJUsage(Proc):
|
|
|
1072
1137
|
plugin_opts = {"report": "file://../reports/tcr/VJUsage.svelte"}
|
|
1073
1138
|
|
|
1074
1139
|
|
|
1140
|
+
@mark(deprecated=True)
|
|
1075
1141
|
class Attach2Seurat(Proc):
|
|
1076
1142
|
"""Attach the clonal information to a Seurat object as metadata
|
|
1077
1143
|
|
|
@@ -1097,10 +1163,10 @@ class Attach2Seurat(Proc):
|
|
|
1097
1163
|
script = "file://../scripts/tcr/Attach2Seurat.R"
|
|
1098
1164
|
|
|
1099
1165
|
|
|
1100
|
-
class
|
|
1101
|
-
"""Cluster the TCR clones by their CDR3 sequences
|
|
1166
|
+
class CDR3Clustering(Proc):
|
|
1167
|
+
"""Cluster the TCR/BCR clones by their CDR3 sequences
|
|
1102
1168
|
|
|
1103
|
-
This process is used to cluster TCR clones based on their CDR3 sequences.
|
|
1169
|
+
This process is used to cluster TCR/BCR clones based on their CDR3 sequences.
|
|
1104
1170
|
|
|
1105
1171
|
It uses either
|
|
1106
1172
|
|
|
@@ -1124,10 +1190,9 @@ class TCRClustering(Proc):
|
|
|
1124
1190
|
yield similar results.
|
|
1125
1191
|
|
|
1126
1192
|
A text file will be generated with the cluster assignments for each cell, together
|
|
1127
|
-
with the `immunarch` object (in `R`) with the cluster assignments at `
|
|
1128
|
-
column. This information will then be merged to
|
|
1129
|
-
|
|
1130
|
-
Futher downstream analysis can be performed using the cluster assignments.
|
|
1193
|
+
with the `immunarch` object (in `R`) with the cluster assignments at `CDR3_Clsuter`
|
|
1194
|
+
column. This information will then be merged to a `Seurat` object for further
|
|
1195
|
+
downstream analysis.
|
|
1131
1196
|
|
|
1132
1197
|
The cluster assignments are prefixed with `S_` or `M_` to indicate whether a
|
|
1133
1198
|
cluster has only one unique CDR3 sequence or multiple CDR3 sequences.
|
|
@@ -1135,17 +1200,20 @@ class TCRClustering(Proc):
|
|
|
1135
1200
|
CDR3 sequence may be shared by multiple cells.
|
|
1136
1201
|
|
|
1137
1202
|
Input:
|
|
1138
|
-
|
|
1203
|
+
screpfile: The TCR/BCR data object loaded by `scRepertoire::CombineTCR()`,
|
|
1204
|
+
`scRepertoire::CombineBCR()` or `scRepertoire::CombineExpression()`
|
|
1139
1205
|
|
|
1140
1206
|
Output:
|
|
1141
|
-
|
|
1142
|
-
|
|
1143
|
-
Columns are CDR3.aa, TCR_Cluster, TCR_Cluster_Size and
|
|
1144
|
-
TCR_Cluster_Size1.
|
|
1145
|
-
TCR_Cluster_Size is the number of cells in the cluster.
|
|
1146
|
-
TCR_Cluster_Size1 is the unique CDR3 sequences in the cluster.
|
|
1207
|
+
outfile: The `scRepertoire` object in qs with TCR/BCR cluster information.
|
|
1208
|
+
Column `CDR3_Cluster` will be added to the metadata.
|
|
1147
1209
|
|
|
1148
1210
|
Envs:
|
|
1211
|
+
type (choice): The type of the data.
|
|
1212
|
+
- TCR: T cell receptor data
|
|
1213
|
+
- BCR: B cell receptor data
|
|
1214
|
+
- auto: Automatically detect the type from the data.
|
|
1215
|
+
Try to find TRB or IGH genes in the CTgene column to determine
|
|
1216
|
+
whether it is TCR or BCR data.
|
|
1149
1217
|
tool (choice): The tool used to do the clustering, either
|
|
1150
1218
|
[GIANA](https://github.com/s175573/GIANA) or
|
|
1151
1219
|
[ClusTCR](https://github.com/svalkiers/clusTCR).
|
|
@@ -1154,34 +1222,51 @@ class TCRClustering(Proc):
|
|
|
1154
1222
|
- ClusTCR: by Sebastiaan Valkiers, etc
|
|
1155
1223
|
python: The path of python with `GIANA`'s dependencies installed
|
|
1156
1224
|
or with `clusTCR` installed. Depending on the `tool` you choose.
|
|
1225
|
+
within_sample (flag): Whether to cluster the TCR/BCR clones within each sample.
|
|
1226
|
+
When `in.screpfile` is a `Seurat` object, the samples are marked by
|
|
1227
|
+
the `Sample` column in the metadata.
|
|
1157
1228
|
args (type=json): The arguments for the clustering tool
|
|
1158
1229
|
For GIANA, they will be passed to `python GIAna.py`
|
|
1159
1230
|
See <https://github.com/s175573/GIANA#usage>.
|
|
1160
1231
|
For ClusTCR, they will be passed to `clustcr.Clustering(...)`
|
|
1161
1232
|
See <https://svalkiers.github.io/clusTCR/docs/clustering/how-to-use.html#clustering>.
|
|
1162
|
-
|
|
1163
|
-
|
|
1233
|
+
chain (choice): The TCR/BCR chain to use for clustering.
|
|
1234
|
+
- heavy: The heavy chain, TRB for TCR, IGH for BCR.
|
|
1235
|
+
For TCR, TRB is the second sequence in `CTaa`, separated by `_` if
|
|
1236
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa2` column.
|
|
1237
|
+
For BCR, IGH is the first sequence in `CTaa`, separated by `_` if
|
|
1238
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa1` column.
|
|
1239
|
+
- light: The light chain, TRA for TCR, IGL/IGK for BCR.
|
|
1240
|
+
For TCR, TRA is the first sequence in `CTaa`, separated by `_` if
|
|
1241
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa1` column.
|
|
1242
|
+
For BCR, IGL/IGK is the second sequence in `CTaa`, separated by `_` if
|
|
1243
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa2` column.
|
|
1244
|
+
- TRA: Only the TRA chain for TCR (light chain).
|
|
1245
|
+
- TRB: Only the TRB chain for TCR (heavy chain).
|
|
1246
|
+
- IGH: Only the IGH chain for BCR (heavy chain).
|
|
1247
|
+
- IGLK: Only the IGL/IGK chain for BCR (light chain).
|
|
1248
|
+
- both: Both sequences from the heavy and light chains (CTaa column).
|
|
1164
1249
|
|
|
1165
1250
|
Requires:
|
|
1166
1251
|
clusTCR:
|
|
1167
1252
|
- if: {{ proc.envs.tool == 'ClusTCR' }}
|
|
1168
1253
|
- check: {{ proc.envs.python }} -c "import clustcr"
|
|
1169
1254
|
""" # noqa: E501
|
|
1170
|
-
input = "
|
|
1171
|
-
output =
|
|
1172
|
-
"immfile:file:{{in.immfile | basename}}",
|
|
1173
|
-
"clusterfile:file:{{in.immfile | stem}}.clusters.txt",
|
|
1174
|
-
]
|
|
1255
|
+
input = "screpfile:file"
|
|
1256
|
+
output = "outfile:file:{{in.screpfile | stem}}.tcr_clustered.qs"
|
|
1175
1257
|
lang = config.lang.rscript
|
|
1176
1258
|
envs = {
|
|
1259
|
+
"type": "auto", # or TCR, BCR
|
|
1177
1260
|
"tool": "GIANA", # or ClusTCR
|
|
1178
|
-
"on_multi": False,
|
|
1179
1261
|
"python": config.lang.python,
|
|
1262
|
+
"within_sample": True, # whether to cluster the TCR clones within each sample
|
|
1180
1263
|
"args": {},
|
|
1264
|
+
"chain": "both",
|
|
1181
1265
|
}
|
|
1182
|
-
script = "file://../scripts/tcr/
|
|
1266
|
+
script = "file://../scripts/tcr/CDR3Clustering.R"
|
|
1183
1267
|
|
|
1184
1268
|
|
|
1269
|
+
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
1185
1270
|
class TCRClusterStats(Proc):
|
|
1186
1271
|
"""Statistics of TCR clusters, generated by `TCRClustering`.
|
|
1187
1272
|
|
|
@@ -1199,7 +1284,7 @@ class TCRClusterStats(Proc):
|
|
|
1199
1284
|
by = "Sample"
|
|
1200
1285
|
```
|
|
1201
1286
|
|
|
1202
|
-
{: width="80%"}
|
|
1287
|
+
{: width="80%"}
|
|
1203
1288
|
|
|
1204
1289
|
### Shared clusters
|
|
1205
1290
|
|
|
@@ -1209,7 +1294,7 @@ class TCRClusterStats(Proc):
|
|
|
1209
1294
|
heatmap_meta = ["region"]
|
|
1210
1295
|
```
|
|
1211
1296
|
|
|
1212
|
-
{: width="80%"}
|
|
1297
|
+
{: width="80%"}
|
|
1213
1298
|
|
|
1214
1299
|
### Sample diversity
|
|
1215
1300
|
|
|
@@ -1218,11 +1303,11 @@ class TCRClusterStats(Proc):
|
|
|
1218
1303
|
method = "gini"
|
|
1219
1304
|
```
|
|
1220
1305
|
|
|
1221
|
-
{: width="80%"}
|
|
1306
|
+
{: width="80%"}
|
|
1222
1307
|
|
|
1223
1308
|
Compared to the sample diversity using TCR clones:
|
|
1224
1309
|
|
|
1225
|
-
{: width="80%"}
|
|
1310
|
+
{: width="80%"}
|
|
1226
1311
|
|
|
1227
1312
|
Input:
|
|
1228
1313
|
immfile: The immunarch object with TCR clusters attached
|
|
@@ -1250,6 +1335,10 @@ class TCRClusterStats(Proc):
|
|
|
1250
1335
|
numbers on the heatmap.
|
|
1251
1336
|
- heatmap_meta (list): The columns of metadata to show on the
|
|
1252
1337
|
heatmap.
|
|
1338
|
+
- cluster_rows (flag): Whether to cluster the rows on the heatmap.
|
|
1339
|
+
- sample_order: The order of the samples on the heatmap.
|
|
1340
|
+
Either a string separated by `,` or a list of sample names.
|
|
1341
|
+
This only works for columns if `cluster_rows` is `True`.
|
|
1253
1342
|
- grouping: The groups to investigate the shared clusters.
|
|
1254
1343
|
If specified, venn diagrams will be drawn instead of heatmaps.
|
|
1255
1344
|
In such case, `numbers_on_heatmap` and `heatmap_meta` will be
|
|
@@ -1313,6 +1402,9 @@ class TCRClusterStats(Proc):
|
|
|
1313
1402
|
"shared_clusters": {
|
|
1314
1403
|
"numbers_on_heatmap": True,
|
|
1315
1404
|
"heatmap_meta": [],
|
|
1405
|
+
"cluster_rows": True,
|
|
1406
|
+
"sample_order": None,
|
|
1407
|
+
"cluster_rows": True,
|
|
1316
1408
|
"grouping": None,
|
|
1317
1409
|
"devpars": {"width": 1000, "height": 1000, "res": 100},
|
|
1318
1410
|
"cases": {},
|
|
@@ -1330,6 +1422,7 @@ class TCRClusterStats(Proc):
|
|
|
1330
1422
|
}
|
|
1331
1423
|
|
|
1332
1424
|
|
|
1425
|
+
@mark(deprecated=True)
|
|
1333
1426
|
class CloneSizeQQPlot(Proc):
|
|
1334
1427
|
"""QQ plot of the clone sizes
|
|
1335
1428
|
|
|
@@ -1389,15 +1482,9 @@ class CDR3AAPhyschem(Proc):
|
|
|
1389
1482
|
- [Zamyatnin, A. A. Protein volume in solution. Prog. Biophys. Mol. Biol. 24, 107-123 (1972).](https://www.sciencedirect.com/science/article/pii/0079610772900053)
|
|
1390
1483
|
|
|
1391
1484
|
Input:
|
|
1392
|
-
|
|
1393
|
-
|
|
1394
|
-
|
|
1395
|
-
It could also be a tab delimited file with `meta.data` of the
|
|
1396
|
-
`Seurat` object.
|
|
1397
|
-
It has to have a `Sample` column, which is used to match the
|
|
1398
|
-
`immdata` object.
|
|
1399
|
-
It is optional, if not provided, the metadata from the `immdata`
|
|
1400
|
-
object will be used.
|
|
1485
|
+
scrfile: The data loaded by `ScRepCombiningExpression`, saved in RDS or qs/qs2 format.
|
|
1486
|
+
The data is actually generated by `scRepertiore::combineExpression()`.
|
|
1487
|
+
The data must have both TRA and TRB chains.
|
|
1401
1488
|
|
|
1402
1489
|
Output:
|
|
1403
1490
|
outdir: The output directory
|
|
@@ -1406,41 +1493,32 @@ class CDR3AAPhyschem(Proc):
|
|
|
1406
1493
|
group: The key of group in metadata to define the groups to
|
|
1407
1494
|
compare. For example, `CellType`, which has cell types annotated
|
|
1408
1495
|
for each cell in the combined object (immdata + Seurat metadata)
|
|
1409
|
-
comparison (type=
|
|
1496
|
+
comparison (type=auto): A dict of two groups, with keys as the
|
|
1410
1497
|
group names and values as the group labels. For example,
|
|
1411
1498
|
```toml
|
|
1412
1499
|
Treg = ["CD4 CTL", "CD4 Naive", "CD4 TCM", "CD4 TEM"]
|
|
1413
1500
|
Tconv = "Tconv"
|
|
1414
1501
|
```
|
|
1415
|
-
|
|
1416
|
-
|
|
1417
|
-
in the `immdata` object separately for each sample. However, the
|
|
1418
|
-
`Seurat` object has a combined `meta.data` for all the samples,
|
|
1419
|
-
so the prefix is needed. Usually, the prefix is the sample name.
|
|
1420
|
-
For example, `Sample1-AACGTTGAGGCTACGT-1`.
|
|
1421
|
-
We need this prefix to add the sample name to the cell names in
|
|
1422
|
-
immdata, so that we can match the cells in `immdata` and
|
|
1423
|
-
`Seurat` object. Set it to `None` or an empty string if the
|
|
1424
|
-
`Seurat` object has the same cell names as `immdata`. You can use
|
|
1425
|
-
placeholders to specify the prefix, e.g., `{Sample}_`. In such a
|
|
1426
|
-
case, the `Sample` column must exist in the `Seurat` object.
|
|
1502
|
+
Or simply a list of two groups, for example, `["Treg", "Tconv"]` when
|
|
1503
|
+
they are both in the `group` column.
|
|
1427
1504
|
target: Which group to use as the target group. The target
|
|
1428
1505
|
group will be labeled as 1, and the other group will be labeled as
|
|
1429
1506
|
0 in the regression.
|
|
1430
|
-
|
|
1431
|
-
|
|
1432
|
-
|
|
1507
|
+
If not specified, the first group in `comparison` will be used as
|
|
1508
|
+
the target group.
|
|
1509
|
+
each (auto): A column, or a list of columns or a string of columns separated by comma.
|
|
1510
|
+
The columns will be used to split the data into multiple groups and the regression will be
|
|
1511
|
+
applied to each group separately.
|
|
1433
1512
|
If not provided, all the cells will be used.
|
|
1434
1513
|
""" # noqa: E501
|
|
1435
|
-
input = "
|
|
1514
|
+
input = "scrfile:file"
|
|
1436
1515
|
output = "outdir:dir:{{in.immdata | stem}}.cdr3aaphyschem"
|
|
1437
1516
|
lang = config.lang.rscript
|
|
1438
1517
|
envs = {
|
|
1439
1518
|
"group": None,
|
|
1440
1519
|
"comparison": None,
|
|
1441
|
-
"prefix": "{Sample}_",
|
|
1442
1520
|
"target": None,
|
|
1443
|
-
"
|
|
1521
|
+
"each": None,
|
|
1444
1522
|
}
|
|
1445
1523
|
script = "file://../scripts/tcr/CDR3AAPhyschem.R"
|
|
1446
1524
|
plugin_opts = {"report": "file://../reports/tcr/CDR3AAPhyschem.svelte"}
|
|
@@ -1480,49 +1558,36 @@ class TESSA(Proc):
|
|
|
1480
1558
|
[link](https://www.nature.com/articles/s42256-021-00383-2)
|
|
1481
1559
|
|
|
1482
1560
|
Input:
|
|
1483
|
-
|
|
1484
|
-
|
|
1485
|
-
|
|
1486
|
-
|
|
1487
|
-
This could also be a tab delimited file (can be gzipped) with
|
|
1488
|
-
expression matrix or dimension reduction results.
|
|
1561
|
+
screpdata: The data loaded by `ScRepCombiningExpression`, saved in RDS or
|
|
1562
|
+
qs/qs2 format.
|
|
1563
|
+
The data is actually generated by `scRepertiore::combineExpression()`.
|
|
1564
|
+
The data must have both TRA and TRB chains.
|
|
1489
1565
|
|
|
1490
1566
|
Output:
|
|
1491
|
-
outfile:
|
|
1492
|
-
(`barcode`, `TESSA_Cluster` and `TESSA_Cluster_Size`) or
|
|
1493
|
-
an RDS file if `in.srtobj` is an RDS file of a Seurat object, with
|
|
1567
|
+
outfile: a qs fileof a Seurat object, with
|
|
1494
1568
|
`TESSA_Cluster` and `TESSA_Cluster_Size` added to the `meta.data`
|
|
1495
1569
|
|
|
1496
1570
|
Envs:
|
|
1497
1571
|
python: The path of python with `TESSA`'s dependencies installed
|
|
1498
|
-
prefix: The prefix to the barcodes of TCR data. You can use placeholder
|
|
1499
|
-
like `{Sample}_` to use the meta data from the immunarch object.
|
|
1500
1572
|
within_sample (flag): Whether the TCR networks are constructed only
|
|
1501
1573
|
within TCRs from the same sample/patient (True) or with all the
|
|
1502
1574
|
TCRs in the meta data matrix (False).
|
|
1503
1575
|
assay: Which assay to use to extract the expression matrix.
|
|
1504
1576
|
Only works if `in.srtobj` is an RDS file of a Seurat object.
|
|
1577
|
+
By default, if `SCTransform` is performed, `SCT` will be used.
|
|
1505
1578
|
predefined_b (flag): Whether use the predefined `b` or not.
|
|
1506
1579
|
Please check the paper of tessa for more details about the b vector.
|
|
1507
1580
|
If True, the tessa will not update b in the MCMC iterations.
|
|
1508
1581
|
max_iter (type=int): The maximum number of iterations for MCMC.
|
|
1509
1582
|
save_tessa (flag): Save tessa detailed results to seurat object?
|
|
1510
|
-
Only works if `in.srtobj` is an RDS file of a Seurat object.
|
|
1511
1583
|
It will be saved to `sobj@misc$tessa`.
|
|
1512
1584
|
"""
|
|
1513
|
-
input = "
|
|
1514
|
-
output = "
|
|
1515
|
-
{%- if in.srtobj.lower().endswith(".rds") -%}
|
|
1516
|
-
{{in.srtobj | stem}}.tessa.RDS
|
|
1517
|
-
{%- else -%}
|
|
1518
|
-
{{in.immdata | stem}}.tessa.txt
|
|
1519
|
-
{%- endif -%}
|
|
1520
|
-
"""
|
|
1585
|
+
input = "screpdata:file"
|
|
1586
|
+
output = "outfile:file:{{in.screpdata | stem}}.tessa.qs"
|
|
1521
1587
|
lang = config.lang.rscript
|
|
1522
1588
|
envs = {
|
|
1523
1589
|
"python": config.lang.python,
|
|
1524
|
-
"
|
|
1525
|
-
"assay": "RNA",
|
|
1590
|
+
"assay": None,
|
|
1526
1591
|
"within_sample": False,
|
|
1527
1592
|
"predefined_b": False,
|
|
1528
1593
|
"max_iter": 1000,
|
|
@@ -1530,3 +1595,523 @@ class TESSA(Proc):
|
|
|
1530
1595
|
}
|
|
1531
1596
|
script = "file://../scripts/tcr/TESSA.R"
|
|
1532
1597
|
plugin_opts = {"report": "file://../reports/tcr/TESSA.svelte"}
|
|
1598
|
+
|
|
1599
|
+
|
|
1600
|
+
class TCRDock(Proc):
|
|
1601
|
+
"""Using TCRDock to predict the structure of MHC-peptide-TCR complexes
|
|
1602
|
+
|
|
1603
|
+
See <https://github.com/phbradley/TCRdock>.
|
|
1604
|
+
|
|
1605
|
+
Input:
|
|
1606
|
+
configfile: The config file for TCRDock
|
|
1607
|
+
It's should be a toml file with the keys listed in `envs`, including
|
|
1608
|
+
`organism`, `mhc_class`, `mhc`, `peptide`, `va`, `ja`, `vb`, `jb`,
|
|
1609
|
+
`cdr3a`, and `cdr3b`.
|
|
1610
|
+
The values will overwrite the values in `envs`.
|
|
1611
|
+
|
|
1612
|
+
Output:
|
|
1613
|
+
outdir: The output directory containing the results
|
|
1614
|
+
|
|
1615
|
+
Envs:
|
|
1616
|
+
organism: The organism of the TCR, peptide and MHC
|
|
1617
|
+
mhc_class (type=int): The MHC class, either `1` or `2`
|
|
1618
|
+
mhc: The MHC allele, e.g., `A*02:01`
|
|
1619
|
+
peptide: The peptide sequence
|
|
1620
|
+
va: The V alpha gene
|
|
1621
|
+
ja: The J alpha gene
|
|
1622
|
+
vb: The V beta gene
|
|
1623
|
+
jb: The J beta gene
|
|
1624
|
+
cdr3a: The CDR3 alpha sequence
|
|
1625
|
+
cdr3b: The CDR3 beta sequence
|
|
1626
|
+
python: The path of python with dependencies for `tcrdock` installed.
|
|
1627
|
+
If not provided, `TCRDock.lang` will be used (the same interpreter
|
|
1628
|
+
used for the wrapper script).
|
|
1629
|
+
It could also be a list to specify, for example, a python in a conda
|
|
1630
|
+
environment (e.g., `["conda", "run", "-n", "myenv", "python"]`).
|
|
1631
|
+
tmpdir: The temporary directory used to clone the `tcrdock` source code if
|
|
1632
|
+
`envs.tcrdock` is not provided.
|
|
1633
|
+
tcrdock: The path to the `tcrdock` source code repo.
|
|
1634
|
+
You need to clone the source code from the github repository.
|
|
1635
|
+
<https://github.com/phbradley/TCRdock> at
|
|
1636
|
+
revision c5a7af42eeb0c2a4492a4d4fe803f1f9aafb6193 at main branch.
|
|
1637
|
+
You also have to run `download_blast.py` after cloning to download the
|
|
1638
|
+
blast database in the directory.
|
|
1639
|
+
If not provided, we will clone the source code to the `envs.tmpdir`
|
|
1640
|
+
directory and run the `download_blast.py` script.
|
|
1641
|
+
model_name: The model name to use
|
|
1642
|
+
model_file: The model file to use.
|
|
1643
|
+
If provided as a relative path, it should be relative to the
|
|
1644
|
+
`<envs.data_dir>/params/`, otherwise, it should be the full path.
|
|
1645
|
+
data_dir: The data directory that contains the model files.
|
|
1646
|
+
The model files should be in the `params` subdirectory.
|
|
1647
|
+
"""
|
|
1648
|
+
input = "configfile:file"
|
|
1649
|
+
output = "outdir:dir:{{in.configfile | stem}}.tcrdock"
|
|
1650
|
+
lang = config.lang.python
|
|
1651
|
+
envs = {
|
|
1652
|
+
"tcrdock": None,
|
|
1653
|
+
"organism": "human",
|
|
1654
|
+
"mhc_class": 1,
|
|
1655
|
+
"mhc": "A*02:01",
|
|
1656
|
+
"peptide": None,
|
|
1657
|
+
"va": None,
|
|
1658
|
+
"ja": None,
|
|
1659
|
+
"vb": None,
|
|
1660
|
+
"jb": None,
|
|
1661
|
+
"cdr3a": None,
|
|
1662
|
+
"cdr3b": None,
|
|
1663
|
+
"python": None,
|
|
1664
|
+
"model_name": "model_2_ptm_ft4",
|
|
1665
|
+
"model_file": "tcrpmhc_run4_af_mhc_params_891.pkl",
|
|
1666
|
+
"data_dir": None,
|
|
1667
|
+
}
|
|
1668
|
+
script = "file://../scripts/tcr/TCRDock.py"
|
|
1669
|
+
|
|
1670
|
+
|
|
1671
|
+
class ScRepLoading(Proc):
|
|
1672
|
+
"""Load the single cell TCR/BCR data into a `scRepertoire` compatible object
|
|
1673
|
+
|
|
1674
|
+
This process loads the single cell TCR/BCR data into a `scRepertoire`
|
|
1675
|
+
(>= v2.0.8, < v2.3.2) compatible object. Later, `scRepertoire::combineExpression`
|
|
1676
|
+
can be used to combine the expression data with the TCR/BCR data.
|
|
1677
|
+
|
|
1678
|
+
For the data path specified at `TCRData`/`BCRData` in the input file
|
|
1679
|
+
(`in.metafile`), will be used to find the TCR/BCR data files and
|
|
1680
|
+
`scRepertoire::loadContigs()` will be used to load the data.
|
|
1681
|
+
|
|
1682
|
+
A directory can be specified in `TCRData`/`BCRData`, then
|
|
1683
|
+
`scRepertoire::loadContigs()` will be used directly to load the data from the
|
|
1684
|
+
directory. Otherwise if a file is specified, it will be symbolically linked to
|
|
1685
|
+
a directory for `scRepertoire::loadContigs()` to load.
|
|
1686
|
+
Note that when the file name can not be recognized by `scRepertoire::loadContigs()`,
|
|
1687
|
+
`envs.format` must be set for the correct format of the data.
|
|
1688
|
+
|
|
1689
|
+
Input:
|
|
1690
|
+
metafile: The meta data of the samples
|
|
1691
|
+
A tab-delimited file
|
|
1692
|
+
Two columns are required:
|
|
1693
|
+
* `Sample` to specify the sample names.
|
|
1694
|
+
* `TCRData`/`BCRData` to assign the path of the data to the samples,
|
|
1695
|
+
and this column will be excluded as metadata.
|
|
1696
|
+
|
|
1697
|
+
Output:
|
|
1698
|
+
outfile: The `scRepertoire` compatible object in qs/qs2 format
|
|
1699
|
+
|
|
1700
|
+
Envs:
|
|
1701
|
+
type (choice): The type of the data to load.
|
|
1702
|
+
- TCR: T cell receptor data
|
|
1703
|
+
- BCR: B cell receptor data
|
|
1704
|
+
- auto: Automatically detect the type from the metadata.
|
|
1705
|
+
If `auto` is selected, the type will be determined by the presence of
|
|
1706
|
+
`TCRData` or `BCRData` columns in the metadata. If both columns are
|
|
1707
|
+
present, `TCR` will be selected by default.
|
|
1708
|
+
combineTCR (type=json): The extra arguments for `scRepertoire::combineTCR`
|
|
1709
|
+
function.
|
|
1710
|
+
See also <https://www.borch.dev/uploads/screpertoire/reference/combinetcr>
|
|
1711
|
+
combineBCR (type=json): The extra arguments for `scRepertoire::combineBCR`
|
|
1712
|
+
function.
|
|
1713
|
+
See also <https://www.borch.dev/uploads/screpertoire/reference/combinebcr>
|
|
1714
|
+
exclude (auto): The columns to exclude from the metadata to add to the object.
|
|
1715
|
+
A list of column names to exclude or a string with column names separated
|
|
1716
|
+
by `,`. By default, `BCRData`, `TCRData` and `RNAData` will be excluded.
|
|
1717
|
+
tmpdir: The temporary directory to store the symbolic links to the
|
|
1718
|
+
TCR/BCR data files.
|
|
1719
|
+
format (choice): The format of the TCR/BCR data files.
|
|
1720
|
+
- 10X: 10X Genomics data, which is usually in a directory with
|
|
1721
|
+
`filtered_contig_annotations.csv` file.
|
|
1722
|
+
- AIRR: AIRR format, which is usually in a file with
|
|
1723
|
+
`airr_rearrangement.tsv` file.
|
|
1724
|
+
- BD: Becton Dickinson data, which is usually in a file with
|
|
1725
|
+
`Contigs_AIRR.tsv` file.
|
|
1726
|
+
- Dandelion: Dandelion data, which is usually in a file with
|
|
1727
|
+
`all_contig_dandelion.tsv` file.
|
|
1728
|
+
- Immcantation: Immcantation data, which is usually in a file with
|
|
1729
|
+
`data.tsv` file.
|
|
1730
|
+
- JSON: JSON format, which is usually in a file with `.json` extension.
|
|
1731
|
+
- ParseBio: ParseBio data, which is usually in a file with
|
|
1732
|
+
`barcode_report.tsv` file.
|
|
1733
|
+
- MiXCR: MiXCR data, which is usually in a file with `clones.tsv` file.
|
|
1734
|
+
- Omniscope: Omniscope data, which is usually in a file with `.csv`
|
|
1735
|
+
extension.
|
|
1736
|
+
- TRUST4: TRUST4 data, which is usually in a file with
|
|
1737
|
+
`barcode_report.tsv` file.
|
|
1738
|
+
- WAT3R: WAT3R data, which is usually in a file with
|
|
1739
|
+
`barcode_results.csv` file.
|
|
1740
|
+
See also: <https://rdrr.io/github/ncborcherding/scRepertoire/man/loadContigs.html>
|
|
1741
|
+
If not provided, the format will be guessed from the file name by `scRepertoire::loadContigs()`.
|
|
1742
|
+
""" # noqa: E501
|
|
1743
|
+
input = "metafile:file"
|
|
1744
|
+
output = "outfile:file:{{in.metafile | stem}}.scRep.qs"
|
|
1745
|
+
lang = config.lang.rscript
|
|
1746
|
+
envs = {
|
|
1747
|
+
"type": "auto", # or TCR/BCR
|
|
1748
|
+
"combineTCR": {"samples": True},
|
|
1749
|
+
"combineBCR": {"samples": True},
|
|
1750
|
+
"exclude": ["BCRData", "TCRData", "RNAData"],
|
|
1751
|
+
"format": None,
|
|
1752
|
+
"tmpdir": config.path.tmpdir,
|
|
1753
|
+
}
|
|
1754
|
+
script = "file://../scripts/tcr/ScRepLoading.R"
|
|
1755
|
+
|
|
1756
|
+
|
|
1757
|
+
class ScRepCombiningExpression(Proc):
|
|
1758
|
+
"""Combine the scTCR/BCR data with the expression data
|
|
1759
|
+
|
|
1760
|
+
This process combines the scTCR/BCR data with the expression data using
|
|
1761
|
+
`scRepertoire::combineExpression` function. The expression data should be
|
|
1762
|
+
in `Seurat` format. The `scRepertoire` object should be a combined contig
|
|
1763
|
+
object, usually generated by `scRepertoire::combineTCR` or
|
|
1764
|
+
`scRepertoire::combineBCR`.
|
|
1765
|
+
|
|
1766
|
+
See also: <https://www.borch.dev/uploads/screpertoire/reference/combineexpression>.
|
|
1767
|
+
|
|
1768
|
+
Input:
|
|
1769
|
+
screpfile: The `scRepertoire` object in RDS/qs format
|
|
1770
|
+
srtobj: The `Seurat` object, saved in RDS/qs format
|
|
1771
|
+
|
|
1772
|
+
Output:
|
|
1773
|
+
outfile: The `Seurat` object with the TCR/BCR data combined
|
|
1774
|
+
In addition to the meta columns added by
|
|
1775
|
+
`scRepertoire::combineExpression()`, a new column `VDJ_Presence` will be
|
|
1776
|
+
added to the metadata. It indicates whether the cell has a TCR/BCR
|
|
1777
|
+
sequence or not. The value is `TRUE` if the cell has a TCR/BCR sequence,
|
|
1778
|
+
and `FALSE` otherwise.
|
|
1779
|
+
|
|
1780
|
+
Envs:
|
|
1781
|
+
cloneCall: How to call the clone - VDJC gene (gene), CDR3 nucleotide (nt),
|
|
1782
|
+
CDR3 amino acid (aa), VDJC gene + CDR3 nucleotide (strict) or
|
|
1783
|
+
a custom variable in the data.
|
|
1784
|
+
chain: indicate if both or a specific chain should be used
|
|
1785
|
+
e.g. "both", "TRA", "TRG", "IGH", "IGL".
|
|
1786
|
+
group_by: The column label in the combined clones in which clone frequency will
|
|
1787
|
+
be calculated. NULL or "none" will keep the format of input.data.
|
|
1788
|
+
proportion (flag): Whether to proportion (TRUE) or total frequency (FALSE) of
|
|
1789
|
+
the clone based on the group_by variable.
|
|
1790
|
+
filterNA (flag): Method to subset Seurat/SCE object of barcodes without clone
|
|
1791
|
+
information
|
|
1792
|
+
cloneSize (type=json): The bins for the grouping based on proportion or
|
|
1793
|
+
frequency.
|
|
1794
|
+
If proportion is FALSE and the cloneSizes are not set high enough based on
|
|
1795
|
+
frequency, the upper limit of cloneSizes will be automatically updated.
|
|
1796
|
+
addLabel (flag): This will add a label to the frequency header, allowing the
|
|
1797
|
+
user to try multiple group_by variables or recalculate frequencies after
|
|
1798
|
+
subsetting the data.
|
|
1799
|
+
"""
|
|
1800
|
+
input = "screpfile:file,srtobj:file"
|
|
1801
|
+
output = "outfile:file:{{in.screpfile | stem}}.qs"
|
|
1802
|
+
lang = config.lang.rscript
|
|
1803
|
+
envs = {
|
|
1804
|
+
"cloneCall": "aa",
|
|
1805
|
+
"chain": "both",
|
|
1806
|
+
"group_by": "Sample",
|
|
1807
|
+
"proportion": True,
|
|
1808
|
+
"filterNA": False,
|
|
1809
|
+
"cloneSize": {
|
|
1810
|
+
"Rare": 1e-04,
|
|
1811
|
+
"Small": 0.001,
|
|
1812
|
+
"Medium": 0.01,
|
|
1813
|
+
"Large": 0.1,
|
|
1814
|
+
"Hyperexpanded": 1,
|
|
1815
|
+
},
|
|
1816
|
+
"addLabel": False,
|
|
1817
|
+
}
|
|
1818
|
+
script = "file://../scripts/tcr/ScRepCombiningExpression.R"
|
|
1819
|
+
|
|
1820
|
+
|
|
1821
|
+
class ClonalStats(Proc):
|
|
1822
|
+
"""Visualize the clonal information.
|
|
1823
|
+
|
|
1824
|
+
Using [`scplotter`](https://github.com/pwwang/scplotter) to visualize the clonal
|
|
1825
|
+
information.
|
|
1826
|
+
|
|
1827
|
+
Examples:
|
|
1828
|
+
### Clonal Volume
|
|
1829
|
+
|
|
1830
|
+
```toml
|
|
1831
|
+
[ClonalStats.envs.cases."Clonal Volume"]
|
|
1832
|
+
viz_type = "volume"
|
|
1833
|
+
x_text_angle = 45
|
|
1834
|
+
```
|
|
1835
|
+
|
|
1836
|
+
{: width="80%"}
|
|
1837
|
+
|
|
1838
|
+
### Clonal Volume by Diagnosis
|
|
1839
|
+
|
|
1840
|
+
```toml
|
|
1841
|
+
[ClonalStats.envs.cases."Clonal Volume by Diagnosis"]
|
|
1842
|
+
viz_type = "volume"
|
|
1843
|
+
x = "seurat_clusters"
|
|
1844
|
+
group_by = "Diagnosis"
|
|
1845
|
+
comparisons = true
|
|
1846
|
+
```
|
|
1847
|
+
|
|
1848
|
+
{: width="80%"}
|
|
1849
|
+
|
|
1850
|
+
### Clonal Abundance
|
|
1851
|
+
|
|
1852
|
+
```toml
|
|
1853
|
+
[ClonalStats.envs.cases."Clonal Abundance"]
|
|
1854
|
+
viz_type = "abundance"
|
|
1855
|
+
```
|
|
1856
|
+
|
|
1857
|
+
{: width="80%"}
|
|
1858
|
+
|
|
1859
|
+
### Clonal Abundance Density
|
|
1860
|
+
|
|
1861
|
+
```toml
|
|
1862
|
+
[ClonalStats.envs.cases."Clonal Abundance Density"]
|
|
1863
|
+
viz_type = "abundance"
|
|
1864
|
+
plot_type = "density"
|
|
1865
|
+
```
|
|
1866
|
+
|
|
1867
|
+
{: width="80%"}
|
|
1868
|
+
|
|
1869
|
+
### CDR3 Length
|
|
1870
|
+
|
|
1871
|
+
```toml
|
|
1872
|
+
[ClonalStats.envs.cases."CDR3 Length"]
|
|
1873
|
+
viz_type = "length"
|
|
1874
|
+
```
|
|
1875
|
+
|
|
1876
|
+
{: width="80%"}
|
|
1877
|
+
|
|
1878
|
+
### CDR3 Length (Beta Chain)
|
|
1879
|
+
|
|
1880
|
+
```toml
|
|
1881
|
+
[ClonalStats.envs.cases."CDR3 Length (Beta Chain)"]
|
|
1882
|
+
viz_type = "length"
|
|
1883
|
+
chain = "TRB"
|
|
1884
|
+
```
|
|
1885
|
+
|
|
1886
|
+
{: width="80%"}
|
|
1887
|
+
|
|
1888
|
+
### Clonal Residency
|
|
1889
|
+
|
|
1890
|
+
```toml
|
|
1891
|
+
[ClonalStats.envs.cases."Clonal Residency"]
|
|
1892
|
+
viz_type = "residency"
|
|
1893
|
+
group_by = "Diagnosis"
|
|
1894
|
+
chain = "TRB"
|
|
1895
|
+
clone_call = "gene"
|
|
1896
|
+
groups = ["Colitis", "NoColitis"]
|
|
1897
|
+
```
|
|
1898
|
+
|
|
1899
|
+
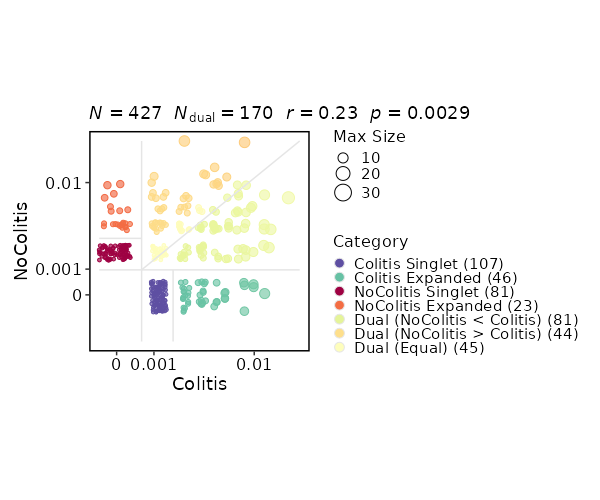{: width="80%"}
|
|
1900
|
+
|
|
1901
|
+
### Clonal Residency (UpSet Plot)
|
|
1902
|
+
|
|
1903
|
+
```toml
|
|
1904
|
+
[ClonalStats.envs.cases."Clonal Residency (UpSet Plot)"]
|
|
1905
|
+
viz_type = "residency"
|
|
1906
|
+
plot_type = "upset"
|
|
1907
|
+
group_by = "Diagnosis"
|
|
1908
|
+
chain = "TRB"
|
|
1909
|
+
clone_call = "gene"
|
|
1910
|
+
groups = ["Colitis", "NoColitis"]
|
|
1911
|
+
devpars = {width = 800}
|
|
1912
|
+
```
|
|
1913
|
+
|
|
1914
|
+
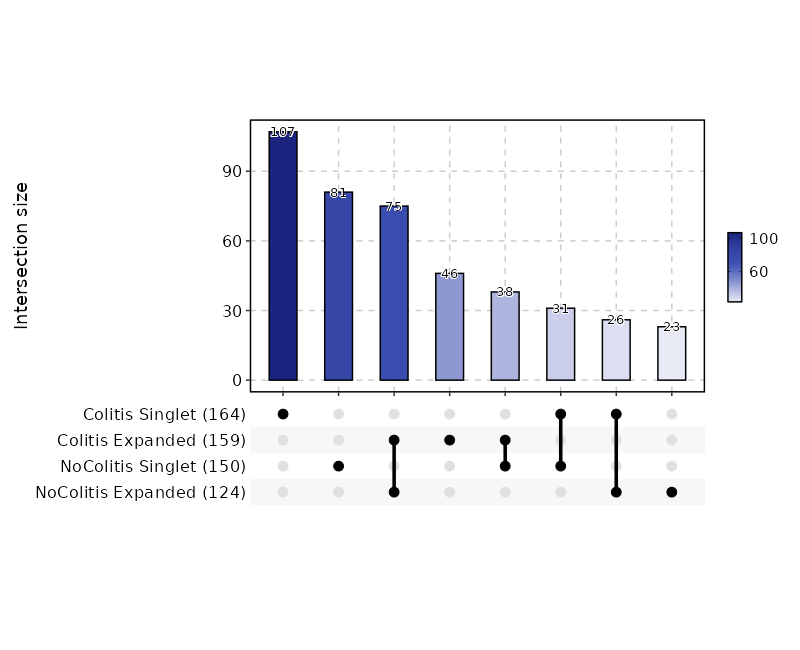{: width="80%"}
|
|
1915
|
+
|
|
1916
|
+
### Clonal Statistics with Expanded Clones
|
|
1917
|
+
|
|
1918
|
+
```toml
|
|
1919
|
+
[ClonalStats.envs.cases."Clonal Statistics with Expanded Clones"]
|
|
1920
|
+
viz_type = "stat"
|
|
1921
|
+
plot_type = "pies"
|
|
1922
|
+
group_by = "Diagnosis"
|
|
1923
|
+
groups = ["Colitis", "NoColitis"]
|
|
1924
|
+
clones = {"Expanded Clones In Colitis" = "sel(Colitis > 2)", "Expanded Clones In NoColitis" = "sel(NoColitis > 2)"}
|
|
1925
|
+
subgroup_by = "seurat_clusters"
|
|
1926
|
+
pie_size = "sqrt"
|
|
1927
|
+
show_row_names = true
|
|
1928
|
+
show_column_names = true
|
|
1929
|
+
devpars = {width = 720}
|
|
1930
|
+
```
|
|
1931
|
+
|
|
1932
|
+
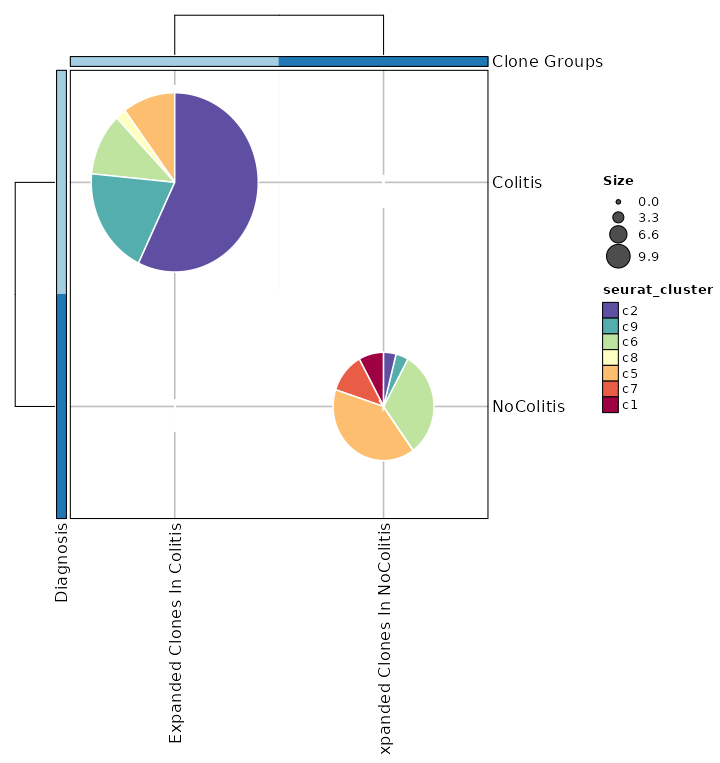{: width="80%"}
|
|
1933
|
+
|
|
1934
|
+
### Hyperexpanded Clonal Dynamics
|
|
1935
|
+
|
|
1936
|
+
```toml
|
|
1937
|
+
[ClonalStats.envs.cases."Hyperexpanded Clonal Dynamics"]
|
|
1938
|
+
viz_type = "stat"
|
|
1939
|
+
plot_type = "sankey"
|
|
1940
|
+
group_by = "Diagnosis"
|
|
1941
|
+
chain = "TRB"
|
|
1942
|
+
groups = ["Colitis", "NoColitis"]
|
|
1943
|
+
clones = {"Hyper-Expanded Clones In Colitis" = "sel(Colitis > 5)", "Hyper-Expanded Clones In NoColitis" = "sel(NoColitis > 5)"}
|
|
1944
|
+
devpars = {width = 800}
|
|
1945
|
+
```
|
|
1946
|
+
|
|
1947
|
+
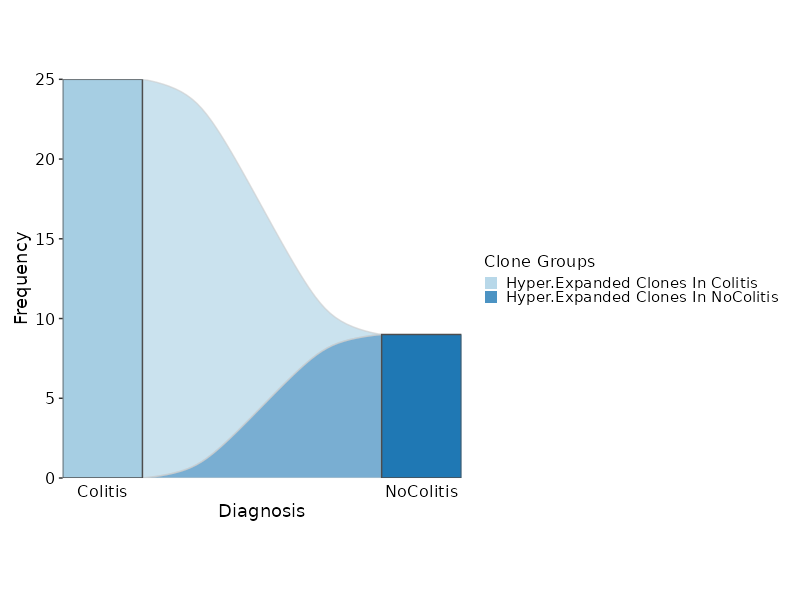{: width="80%"}
|
|
1948
|
+
|
|
1949
|
+
### Clonal Composition
|
|
1950
|
+
|
|
1951
|
+
```toml
|
|
1952
|
+
[ClonalStats.envs.cases."Clonal Composition"]
|
|
1953
|
+
viz_type = "composition"
|
|
1954
|
+
x_text_angle = 45
|
|
1955
|
+
```
|
|
1956
|
+
|
|
1957
|
+
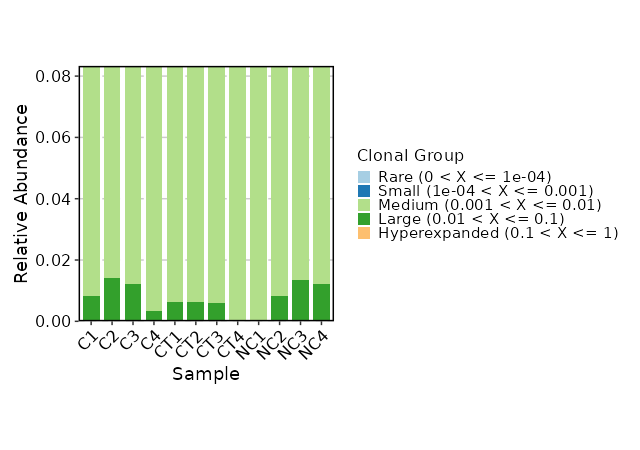{: width="80%"}
|
|
1958
|
+
|
|
1959
|
+
### Clonal Overlapping
|
|
1960
|
+
|
|
1961
|
+
```toml
|
|
1962
|
+
viz_type = "overlap"
|
|
1963
|
+
chain = "TRB"
|
|
1964
|
+
clone_call = "gene"
|
|
1965
|
+
```
|
|
1966
|
+
|
|
1967
|
+
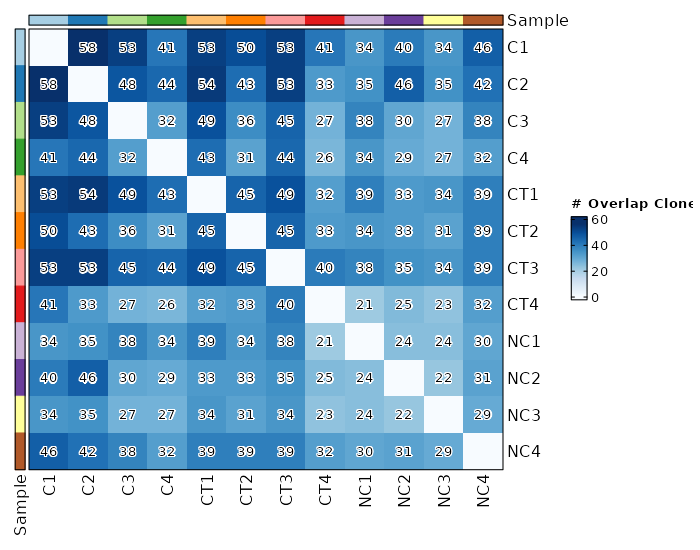{: width="80%"}
|
|
1968
|
+
|
|
1969
|
+
### Clonal Diversity
|
|
1970
|
+
|
|
1971
|
+
```toml
|
|
1972
|
+
[ClonalStats.envs.cases."Clonal Diversity"]
|
|
1973
|
+
# method = "shannon" # default
|
|
1974
|
+
viz_type = "diversity"
|
|
1975
|
+
x_text_angle = 45
|
|
1976
|
+
```
|
|
1977
|
+
|
|
1978
|
+
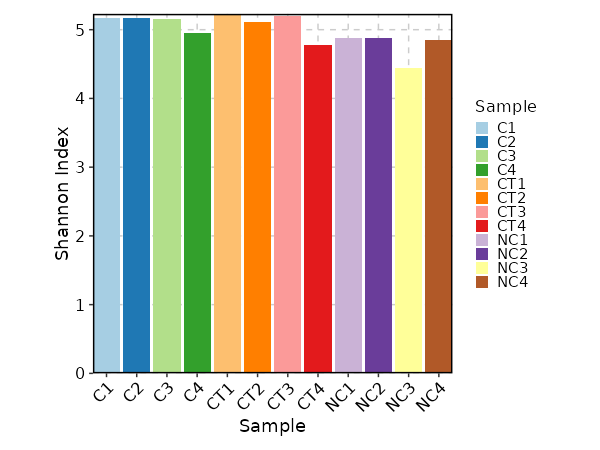{: width="80%"}
|
|
1979
|
+
|
|
1980
|
+
### Clonal Diversity (gini.coeff, by Diagnosis)
|
|
1981
|
+
|
|
1982
|
+
```toml
|
|
1983
|
+
[ClonalStats.envs.cases."Clonal Diversity (gini.coeff, by Diagnosis)"]
|
|
1984
|
+
method = "gini.coeff"
|
|
1985
|
+
viz_type = "diversity"
|
|
1986
|
+
plot_type = "box"
|
|
1987
|
+
group_by = "Diagnosis"
|
|
1988
|
+
comparisons = true
|
|
1989
|
+
devpars = {height = 600, width = 600}
|
|
1990
|
+
```
|
|
1991
|
+
|
|
1992
|
+
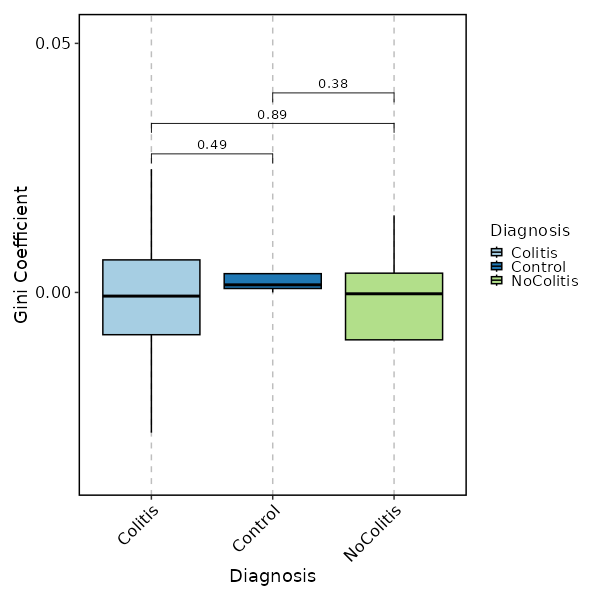{: width="80%"}
|
|
1993
|
+
|
|
1994
|
+
### Gene Usage Frequency
|
|
1995
|
+
|
|
1996
|
+
```toml
|
|
1997
|
+
[ClonalStats.envs.cases."Gene Usage Frequency"]
|
|
1998
|
+
viz_type = "geneusage"
|
|
1999
|
+
devpars = {width = 1200}
|
|
2000
|
+
```
|
|
2001
|
+
|
|
2002
|
+
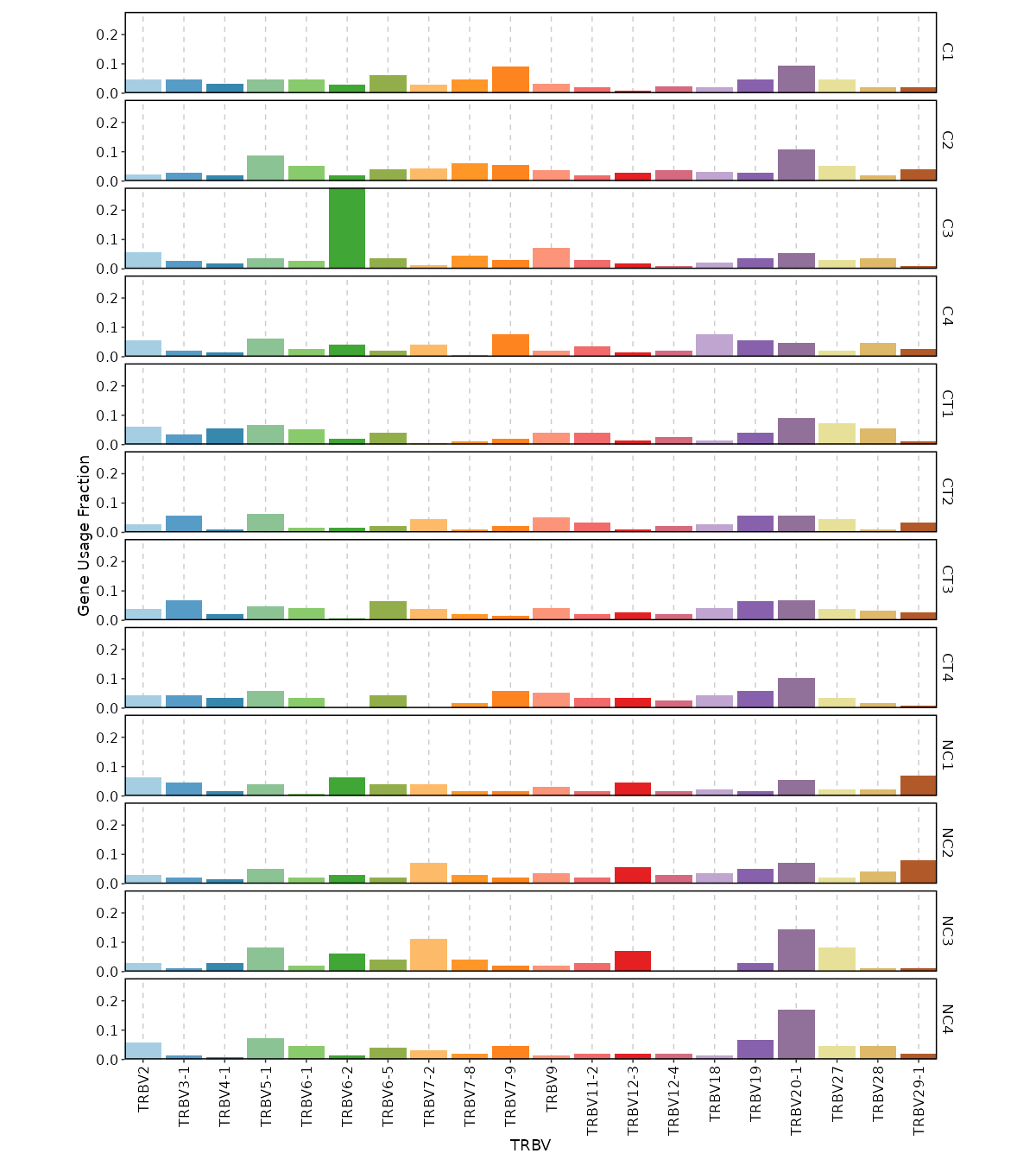{: width="80%"}
|
|
2003
|
+
|
|
2004
|
+
### Positional amino acid frequency
|
|
2005
|
+
|
|
2006
|
+
```toml
|
|
2007
|
+
[ClonalStats.envs.cases."Positional amino acid frequency"]
|
|
2008
|
+
viz_type = "positional"
|
|
2009
|
+
# method = "AA" # default
|
|
2010
|
+
devpars = {width = 1600}
|
|
2011
|
+
```
|
|
2012
|
+
|
|
2013
|
+
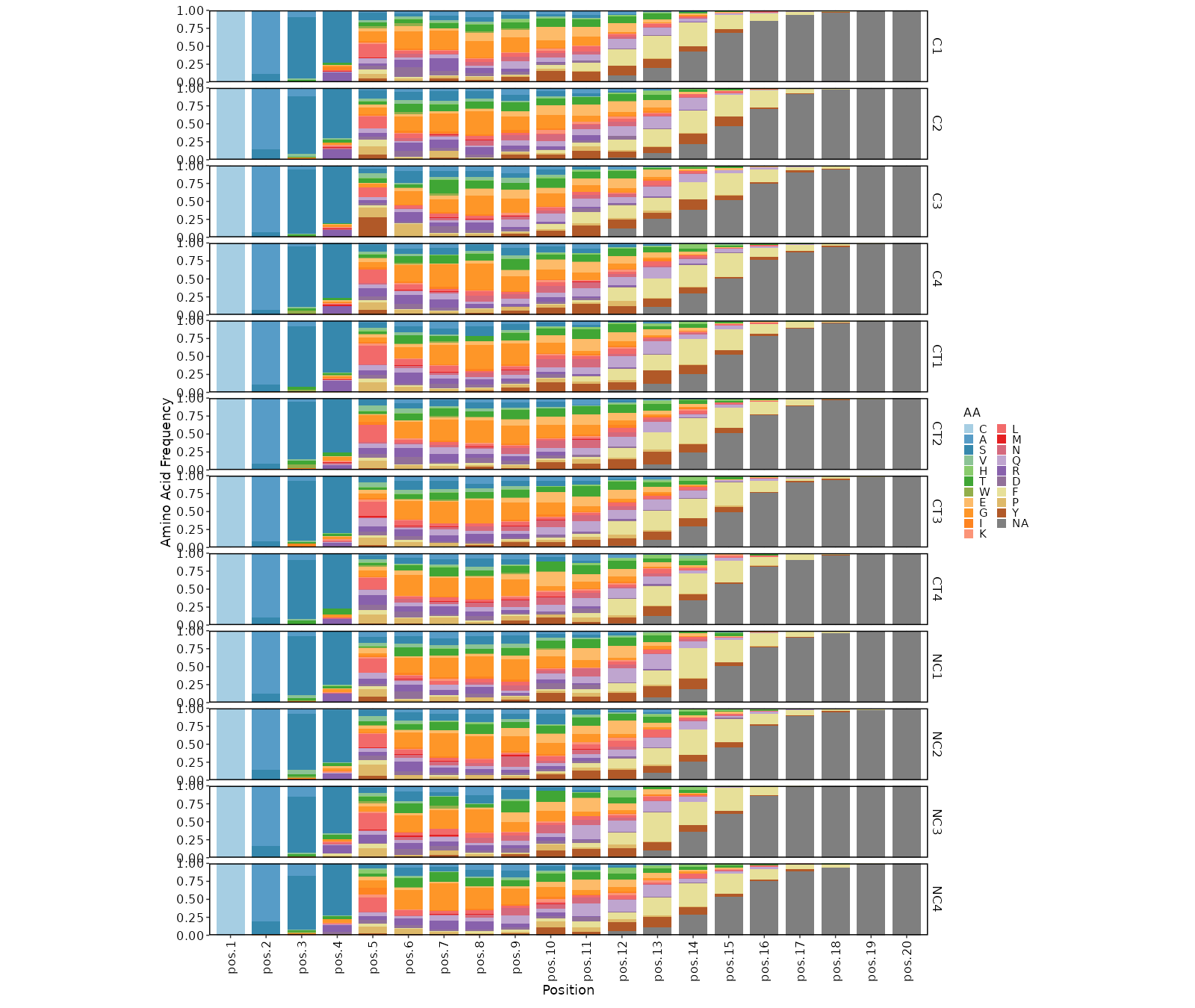{: width="80%"}
|
|
2014
|
+
|
|
2015
|
+
### Positional shannon entropy
|
|
2016
|
+
|
|
2017
|
+
```toml
|
|
2018
|
+
[ClonalStats.envs.cases."Positional shannon entropy"]
|
|
2019
|
+
viz_type = "positional"
|
|
2020
|
+
method = "shannon"
|
|
2021
|
+
devpars = {width = 1200}
|
|
2022
|
+
```
|
|
2023
|
+
|
|
2024
|
+
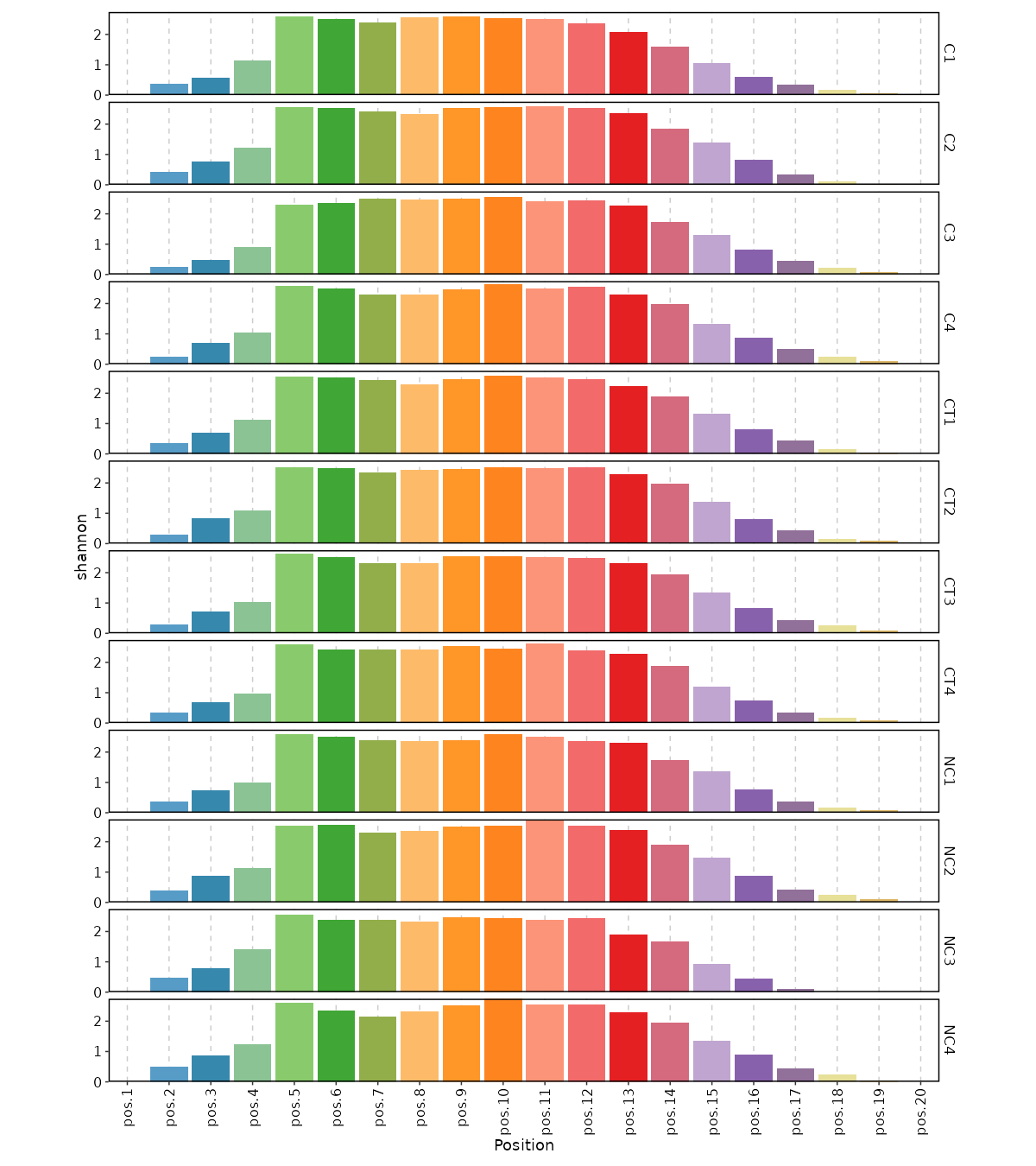{: width="80%"}
|
|
2025
|
+
|
|
2026
|
+
### 3-Mer Frequency
|
|
2027
|
+
|
|
2028
|
+
```toml
|
|
2029
|
+
[ClonalStats.envs.cases."3-Mer Frequency"]
|
|
2030
|
+
viz_type = "kmer"
|
|
2031
|
+
k = 3 # default is 3
|
|
2032
|
+
devpars = {width = 800}
|
|
2033
|
+
```
|
|
2034
|
+
|
|
2035
|
+
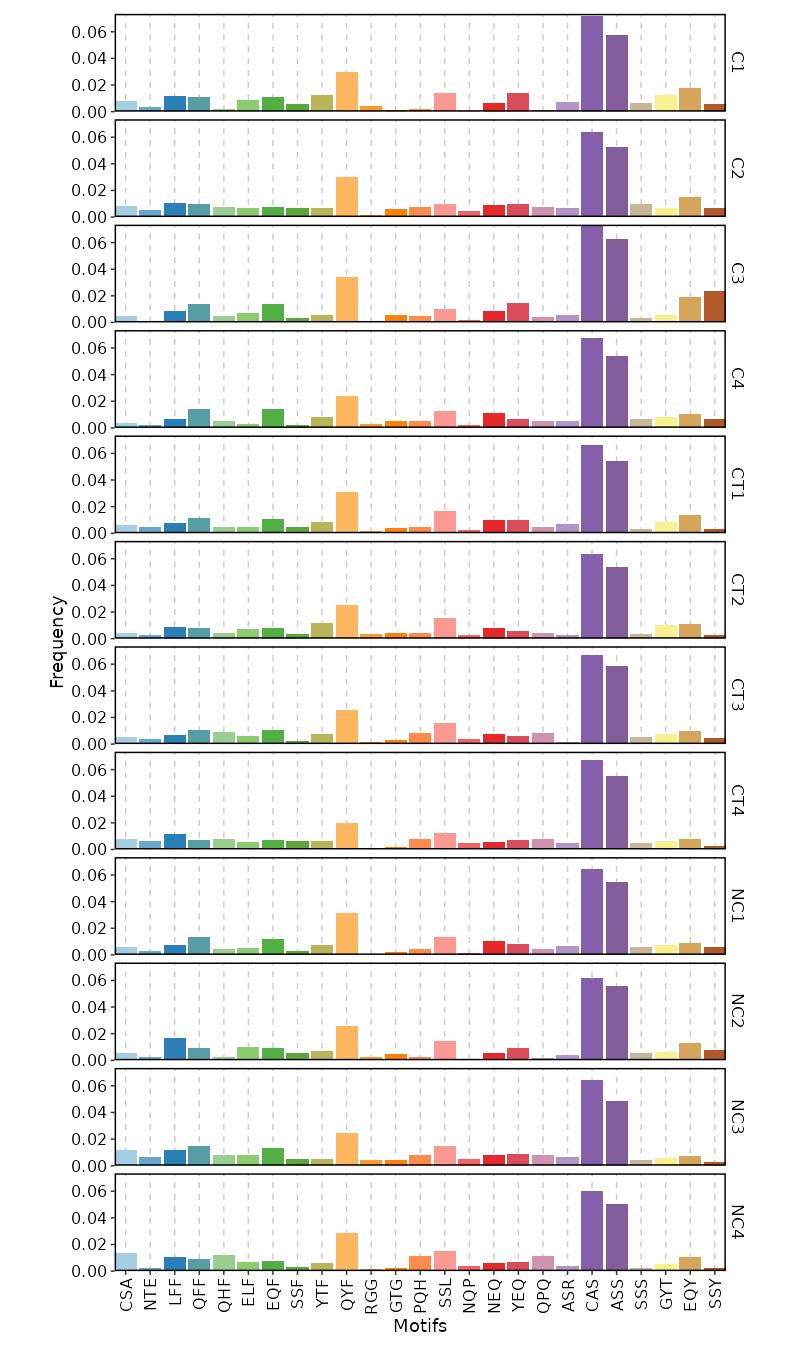{: width="80%"}
|
|
2036
|
+
|
|
2037
|
+
### Rarefaction Curve
|
|
2038
|
+
|
|
2039
|
+
```toml
|
|
2040
|
+
[ClonalStats.envs.cases."Rarefaction Curve"]
|
|
2041
|
+
viz_type = "rarefaction"
|
|
2042
|
+
```
|
|
2043
|
+
|
|
2044
|
+
{: width="80%"}
|
|
2045
|
+
|
|
2046
|
+
Input:
|
|
2047
|
+
screpfile: The `scRepertoire` object in RDS/qs format
|
|
2048
|
+
|
|
2049
|
+
Output:
|
|
2050
|
+
outdir: The output directory containing the plots
|
|
2051
|
+
|
|
2052
|
+
Envs:
|
|
2053
|
+
mutaters (type=json;order=-9): The mutaters passed to `dplyr::mutate()` to add new variables.
|
|
2054
|
+
When the object loaded form `in.screpfile` is a list, the mutaters will be applied to each element.
|
|
2055
|
+
The keys are the names of the new variables, and the values are the expressions.
|
|
2056
|
+
When it is a `Seurat` object, typically an output of `scRepertoire::combineExpression()`,
|
|
2057
|
+
the mutaters will be applied to the `meta.data`.
|
|
2058
|
+
viz_type (choice): The type of visualization to generate.
|
|
2059
|
+
- volume: The volume of the clones using [`ClonalVolumePlot`](https://pwwang.github.io/scplotter/reference/ClonalVolumePlot.html)
|
|
2060
|
+
- abundance: The abundance of the clones using [`ClonalAbundancePlot`](https://pwwang.github.io/scplotter/reference/ClonalAbundancePlot.html)
|
|
2061
|
+
- length: The length of the CDR3 sequences using [`ClonalLengthPlot`](https://pwwang.github.io/scplotter/reference/ClonalLengthPlot.html)
|
|
2062
|
+
- residency: The residency of the clones using [`ClonalResidencyPlot`](https://pwwang.github.io/scplotter/reference/ClonalResidencyPlot.html)
|
|
2063
|
+
- stats: The stats of the clones using [`ClonalStatsPlot`](https://pwwang.github.io/scplotter/reference/ClonalStatsPlot.html)
|
|
2064
|
+
- composition: The composition of the clones using [`ClonalCompositionPlot`](https://pwwang.github.io/scplotter/reference/ClonalCompositionPlot.html)
|
|
2065
|
+
- overlap: The overlap of the clones using [`ClonalOverlapPlot`](https://pwwang.github.io/scplotter/reference/ClonalOverlapPlot.html)
|
|
2066
|
+
- diversity: The diversity of the clones using [`ClonalDiversityPlot`](https://pwwang.github.io/scplotter/reference/ClonalDiversityPlot.html)
|
|
2067
|
+
- geneusage: The gene usage of the clones using [`ClonalGeneUsagePlot`](https://pwwang.github.io/scplotter/reference/ClonalGeneUsagePlot.html)
|
|
2068
|
+
- positional: The positional information of the clones using [`ClonalPositionalPlot`](https://pwwang.github.io/scplotter/reference/ClonalPositionalPlot.html)
|
|
2069
|
+
- kmer: The kmer information of the clones using [`ClonalKmerPlot`](https://pwwang.github.io/scplotter/reference/ClonalKmerPlot.html)
|
|
2070
|
+
- rarefaction: The rarefaction curve of the clones using [`ClonalRarefactionPlot`](https://pwwang.github.io/scplotter/reference/ClonalRarefactionPlot.html)
|
|
2071
|
+
subset: An expression to subset the data before plotting.
|
|
2072
|
+
Similar to `mutaters`, it will be applied to each element by `dplyr::filter()` if the object
|
|
2073
|
+
loaded form `in.screpfile` is a list; otherwise, it will be applied to
|
|
2074
|
+
`subset(sobj, subset = <expr>)` if the object is a `Seurat` object.
|
|
2075
|
+
devpars (ns): The parameters for the plotting device.
|
|
2076
|
+
- width (type=int): The width of the device
|
|
2077
|
+
- height (type=int): The height of the device
|
|
2078
|
+
- res (type=int): The resolution of the device
|
|
2079
|
+
more_formats (list): The extra formats to save the plots in, other than PNG.
|
|
2080
|
+
save_code (flag): Whether to save the code used to generate the plots
|
|
2081
|
+
Note that the data directly used to generate the plots will also be saved in an `rda` file.
|
|
2082
|
+
Be careful if the data is large as it may take a lot of disk space.
|
|
2083
|
+
save_data (flag): Whether to save the data used to generate the plot.
|
|
2084
|
+
descr: The description of the plot, used to show in the report.
|
|
2085
|
+
<more>: The arguments for the plot function
|
|
2086
|
+
See the documentation of the corresponding plot function for the details
|
|
2087
|
+
cases (type=json): The cases to generate the plots if we have multiple cases.
|
|
2088
|
+
The keys are the names of the cases, and the values are the arguments for the plot function.
|
|
2089
|
+
The arguments in `envs` will be used if not specified in `cases`, except for `mutaters`.
|
|
2090
|
+
Sections can be specified as the prefix of the case name, separated by `::`.
|
|
2091
|
+
For example, if you have a case named `Clonal Volume::Case1`, the plot will be put in the
|
|
2092
|
+
section `Clonal Volume`. By default, when there are multiple cases for the same 'viz_type', the name of the 'viz_type' will be used
|
|
2093
|
+
as the default section name (for example, when 'viz_type' is 'volume', the section name will be 'Clonal Volume').
|
|
2094
|
+
When there is only a single case, the section name will default to 'DEFAULT', which will not be shown
|
|
2095
|
+
in the report.
|
|
2096
|
+
""" # noqa: E501
|
|
2097
|
+
input = "screpfile:file"
|
|
2098
|
+
output = "outdir:dir:{{in.screpfile | stem}}.clonalstats"
|
|
2099
|
+
lang = config.lang.rscript
|
|
2100
|
+
envs = {
|
|
2101
|
+
"mutaters": {},
|
|
2102
|
+
"subset": None,
|
|
2103
|
+
"viz_type": None,
|
|
2104
|
+
"devpars": {"width": None, "height": None, "res": 100},
|
|
2105
|
+
"more_formats": [],
|
|
2106
|
+
"save_code": False,
|
|
2107
|
+
"save_data": False,
|
|
2108
|
+
"descr": None,
|
|
2109
|
+
"cases": {
|
|
2110
|
+
"Clonal Volume": {"viz_type": "volume"},
|
|
2111
|
+
"Clonal Abundance": {"viz_type": "abundance"},
|
|
2112
|
+
"CDR3 Length": {"viz_type": "length"},
|
|
2113
|
+
"Clonal Diversity": {"viz_type": "diversity"},
|
|
2114
|
+
}
|
|
2115
|
+
}
|
|
2116
|
+
script = "file://../scripts/tcr/ClonalStats.R"
|
|
2117
|
+
plugin_opts = {"report": "file://../reports/tcr/ClonalStats.svelte"}
|