biopipen 0.34.6__py3-none-any.whl → 0.34.26__py3-none-any.whl
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- biopipen/__init__.py +1 -1
- biopipen/core/config.toml +4 -0
- biopipen/core/filters.py +1 -1
- biopipen/core/testing.py +2 -1
- biopipen/ns/cellranger.py +33 -3
- biopipen/ns/regulatory.py +4 -0
- biopipen/ns/scrna.py +548 -98
- biopipen/ns/scrna_metabolic_landscape.py +4 -0
- biopipen/ns/tcr.py +256 -16
- biopipen/ns/web.py +5 -0
- biopipen/reports/scrna_metabolic_landscape/MetabolicFeatures.svelte +9 -9
- biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayActivity.svelte +9 -8
- biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayHeterogeneity.svelte +9 -9
- biopipen/reports/tcr/ClonalStats.svelte +1 -0
- biopipen/scripts/cellranger/CellRangerCount.py +55 -11
- biopipen/scripts/cellranger/CellRangerVdj.py +54 -8
- biopipen/scripts/regulatory/MotifAffinityTest.R +21 -5
- biopipen/scripts/regulatory/MotifAffinityTest_AtSNP.R +9 -2
- biopipen/scripts/regulatory/MotifAffinityTest_MotifBreakR.R +15 -6
- biopipen/scripts/regulatory/VariantMotifPlot.R +1 -1
- biopipen/scripts/regulatory/motifs-common.R +3 -2
- biopipen/scripts/scrna/AnnData2Seurat.R +2 -1
- biopipen/scripts/scrna/CellCellCommunication.py +26 -14
- biopipen/scripts/scrna/CellCellCommunicationPlots.R +23 -4
- biopipen/scripts/scrna/CellSNPLite.py +30 -0
- biopipen/scripts/scrna/CellTypeAnnotation-celltypist.R +27 -36
- biopipen/scripts/scrna/CellTypeAnnotation-direct.R +42 -26
- biopipen/scripts/scrna/CellTypeAnnotation-hitype.R +11 -13
- biopipen/scripts/scrna/CellTypeAnnotation-sccatch.R +5 -8
- biopipen/scripts/scrna/CellTypeAnnotation-sctype.R +5 -8
- biopipen/scripts/scrna/CellTypeAnnotation.R +26 -3
- biopipen/scripts/scrna/MQuad.py +25 -0
- biopipen/scripts/scrna/MarkersFinder.R +128 -30
- biopipen/scripts/scrna/ModuleScoreCalculator.R +9 -1
- biopipen/scripts/scrna/PseudoBulkDEG.R +113 -27
- biopipen/scripts/scrna/ScFGSEA.R +23 -26
- biopipen/scripts/scrna/ScVelo.py +20 -8
- biopipen/scripts/scrna/SeuratClusterStats-clustree.R +1 -1
- biopipen/scripts/scrna/SeuratClusterStats-features.R +6 -1
- biopipen/scripts/scrna/SeuratClustering.R +5 -1
- biopipen/scripts/scrna/SeuratMap2Ref.R +1 -2
- biopipen/scripts/scrna/SeuratPreparing.R +19 -11
- biopipen/scripts/scrna/SeuratSubClustering.R +1 -1
- biopipen/scripts/scrna/Slingshot.R +2 -4
- biopipen/scripts/scrna/TopExpressingGenes.R +1 -4
- biopipen/scripts/scrna/celltypist-wrapper.py +140 -4
- biopipen/scripts/scrna/scvelo_paga.py +313 -0
- biopipen/scripts/scrna/seurat_anndata_conversion.py +18 -1
- biopipen/scripts/tcr/{TCRClustering.R → CDR3Clustering.R} +63 -23
- biopipen/scripts/tcr/ClonalStats.R +76 -35
- biopipen/utils/misc.py +104 -9
- {biopipen-0.34.6.dist-info → biopipen-0.34.26.dist-info}/METADATA +5 -2
- {biopipen-0.34.6.dist-info → biopipen-0.34.26.dist-info}/RECORD +55 -53
- {biopipen-0.34.6.dist-info → biopipen-0.34.26.dist-info}/WHEEL +1 -1
- biopipen/utils/common_docstrs.py +0 -103
- {biopipen-0.34.6.dist-info → biopipen-0.34.26.dist-info}/entry_points.txt +0 -0
|
@@ -29,6 +29,10 @@ class MetabolicPathwayActivity(Proc):
|
|
|
29
29
|
|
|
30
30
|
{: width="45%"}
|
|
31
31
|
|
|
32
|
+
You may also have a merged heatmap to show all subsets in one plot.
|
|
33
|
+
|
|
34
|
+
{: width="80%"}
|
|
35
|
+
|
|
32
36
|
Input:
|
|
33
37
|
sobjfile: The Seurat object file.
|
|
34
38
|
It should be loaded as a Seurat object
|
biopipen/ns/tcr.py
CHANGED
|
@@ -1163,10 +1163,10 @@ class Attach2Seurat(Proc):
|
|
|
1163
1163
|
script = "file://../scripts/tcr/Attach2Seurat.R"
|
|
1164
1164
|
|
|
1165
1165
|
|
|
1166
|
-
class
|
|
1167
|
-
"""Cluster the TCR clones by their CDR3 sequences
|
|
1166
|
+
class CDR3Clustering(Proc):
|
|
1167
|
+
"""Cluster the TCR/BCR clones by their CDR3 sequences
|
|
1168
1168
|
|
|
1169
|
-
This process is used to cluster TCR clones based on their CDR3 sequences.
|
|
1169
|
+
This process is used to cluster TCR/BCR clones based on their CDR3 sequences.
|
|
1170
1170
|
|
|
1171
1171
|
It uses either
|
|
1172
1172
|
|
|
@@ -1190,7 +1190,7 @@ class TCRClustering(Proc):
|
|
|
1190
1190
|
yield similar results.
|
|
1191
1191
|
|
|
1192
1192
|
A text file will be generated with the cluster assignments for each cell, together
|
|
1193
|
-
with the `immunarch` object (in `R`) with the cluster assignments at `
|
|
1193
|
+
with the `immunarch` object (in `R`) with the cluster assignments at `CDR3_Clsuter`
|
|
1194
1194
|
column. This information will then be merged to a `Seurat` object for further
|
|
1195
1195
|
downstream analysis.
|
|
1196
1196
|
|
|
@@ -1200,14 +1200,20 @@ class TCRClustering(Proc):
|
|
|
1200
1200
|
CDR3 sequence may be shared by multiple cells.
|
|
1201
1201
|
|
|
1202
1202
|
Input:
|
|
1203
|
-
screpfile: The TCR data object loaded by `scRepertoire::CombineTCR()
|
|
1204
|
-
`scRepertoire::CombineExpression()`
|
|
1203
|
+
screpfile: The TCR/BCR data object loaded by `scRepertoire::CombineTCR()`,
|
|
1204
|
+
`scRepertoire::CombineBCR()` or `scRepertoire::CombineExpression()`
|
|
1205
1205
|
|
|
1206
1206
|
Output:
|
|
1207
|
-
outfile: The `scRepertoire` object in qs with TCR cluster information.
|
|
1208
|
-
Column `
|
|
1207
|
+
outfile: The `scRepertoire` object in qs with TCR/BCR cluster information.
|
|
1208
|
+
Column `CDR3_Cluster` will be added to the metadata.
|
|
1209
1209
|
|
|
1210
1210
|
Envs:
|
|
1211
|
+
type (choice): The type of the data.
|
|
1212
|
+
- TCR: T cell receptor data
|
|
1213
|
+
- BCR: B cell receptor data
|
|
1214
|
+
- auto: Automatically detect the type from the data.
|
|
1215
|
+
Try to find TRB or IGH genes in the CTgene column to determine
|
|
1216
|
+
whether it is TCR or BCR data.
|
|
1211
1217
|
tool (choice): The tool used to do the clustering, either
|
|
1212
1218
|
[GIANA](https://github.com/s175573/GIANA) or
|
|
1213
1219
|
[ClusTCR](https://github.com/svalkiers/clusTCR).
|
|
@@ -1216,7 +1222,7 @@ class TCRClustering(Proc):
|
|
|
1216
1222
|
- ClusTCR: by Sebastiaan Valkiers, etc
|
|
1217
1223
|
python: The path of python with `GIANA`'s dependencies installed
|
|
1218
1224
|
or with `clusTCR` installed. Depending on the `tool` you choose.
|
|
1219
|
-
within_sample (flag): Whether to cluster the TCR clones within each sample.
|
|
1225
|
+
within_sample (flag): Whether to cluster the TCR/BCR clones within each sample.
|
|
1220
1226
|
When `in.screpfile` is a `Seurat` object, the samples are marked by
|
|
1221
1227
|
the `Sample` column in the metadata.
|
|
1222
1228
|
args (type=json): The arguments for the clustering tool
|
|
@@ -1224,10 +1230,22 @@ class TCRClustering(Proc):
|
|
|
1224
1230
|
See <https://github.com/s175573/GIANA#usage>.
|
|
1225
1231
|
For ClusTCR, they will be passed to `clustcr.Clustering(...)`
|
|
1226
1232
|
See <https://svalkiers.github.io/clusTCR/docs/clustering/how-to-use.html#clustering>.
|
|
1227
|
-
chain (choice): The TCR chain to use for clustering.
|
|
1228
|
-
-
|
|
1229
|
-
|
|
1230
|
-
|
|
1233
|
+
chain (choice): The TCR/BCR chain to use for clustering.
|
|
1234
|
+
- heavy: The heavy chain, TRB for TCR, IGH for BCR.
|
|
1235
|
+
For TCR, TRB is the second sequence in `CTaa`, separated by `_` if
|
|
1236
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa2` column.
|
|
1237
|
+
For BCR, IGH is the first sequence in `CTaa`, separated by `_` if
|
|
1238
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa1` column.
|
|
1239
|
+
- light: The light chain, TRA for TCR, IGL/IGK for BCR.
|
|
1240
|
+
For TCR, TRA is the first sequence in `CTaa`, separated by `_` if
|
|
1241
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa1` column.
|
|
1242
|
+
For BCR, IGL/IGK is the second sequence in `CTaa`, separated by `_` if
|
|
1243
|
+
input is a Seurat object; otherwise, it is extracted from the `cdr3_aa2` column.
|
|
1244
|
+
- TRA: Only the TRA chain for TCR (light chain).
|
|
1245
|
+
- TRB: Only the TRB chain for TCR (heavy chain).
|
|
1246
|
+
- IGH: Only the IGH chain for BCR (heavy chain).
|
|
1247
|
+
- IGLK: Only the IGL/IGK chain for BCR (light chain).
|
|
1248
|
+
- both: Both sequences from the heavy and light chains (CTaa column).
|
|
1231
1249
|
|
|
1232
1250
|
Requires:
|
|
1233
1251
|
clusTCR:
|
|
@@ -1238,13 +1256,14 @@ class TCRClustering(Proc):
|
|
|
1238
1256
|
output = "outfile:file:{{in.screpfile | stem}}.tcr_clustered.qs"
|
|
1239
1257
|
lang = config.lang.rscript
|
|
1240
1258
|
envs = {
|
|
1259
|
+
"type": "auto", # or TCR, BCR
|
|
1241
1260
|
"tool": "GIANA", # or ClusTCR
|
|
1242
1261
|
"python": config.lang.python,
|
|
1243
1262
|
"within_sample": True, # whether to cluster the TCR clones within each sample
|
|
1244
1263
|
"args": {},
|
|
1245
|
-
"chain": "both",
|
|
1264
|
+
"chain": "both",
|
|
1246
1265
|
}
|
|
1247
|
-
script = "file://../scripts/tcr/
|
|
1266
|
+
script = "file://../scripts/tcr/CDR3Clustering.R"
|
|
1248
1267
|
|
|
1249
1268
|
|
|
1250
1269
|
@mark(deprecated="{proc.name} is deprecated, use ClonalStats instead.")
|
|
@@ -1805,6 +1824,225 @@ class ClonalStats(Proc):
|
|
|
1805
1824
|
Using [`scplotter`](https://github.com/pwwang/scplotter) to visualize the clonal
|
|
1806
1825
|
information.
|
|
1807
1826
|
|
|
1827
|
+
Examples:
|
|
1828
|
+
### Clonal Volume
|
|
1829
|
+
|
|
1830
|
+
```toml
|
|
1831
|
+
[ClonalStats.envs.cases."Clonal Volume"]
|
|
1832
|
+
viz_type = "volume"
|
|
1833
|
+
x_text_angle = 45
|
|
1834
|
+
```
|
|
1835
|
+
|
|
1836
|
+
{: width="80%"}
|
|
1837
|
+
|
|
1838
|
+
### Clonal Volume by Diagnosis
|
|
1839
|
+
|
|
1840
|
+
```toml
|
|
1841
|
+
[ClonalStats.envs.cases."Clonal Volume by Diagnosis"]
|
|
1842
|
+
viz_type = "volume"
|
|
1843
|
+
x = "seurat_clusters"
|
|
1844
|
+
group_by = "Diagnosis"
|
|
1845
|
+
comparisons = true
|
|
1846
|
+
```
|
|
1847
|
+
|
|
1848
|
+
{: width="80%"}
|
|
1849
|
+
|
|
1850
|
+
### Clonal Abundance
|
|
1851
|
+
|
|
1852
|
+
```toml
|
|
1853
|
+
[ClonalStats.envs.cases."Clonal Abundance"]
|
|
1854
|
+
viz_type = "abundance"
|
|
1855
|
+
```
|
|
1856
|
+
|
|
1857
|
+
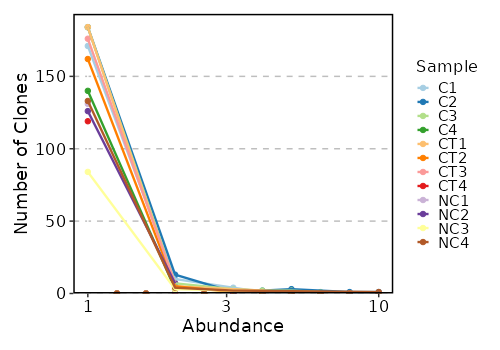{: width="80%"}
|
|
1858
|
+
|
|
1859
|
+
### Clonal Abundance Density
|
|
1860
|
+
|
|
1861
|
+
```toml
|
|
1862
|
+
[ClonalStats.envs.cases."Clonal Abundance Density"]
|
|
1863
|
+
viz_type = "abundance"
|
|
1864
|
+
plot_type = "density"
|
|
1865
|
+
```
|
|
1866
|
+
|
|
1867
|
+
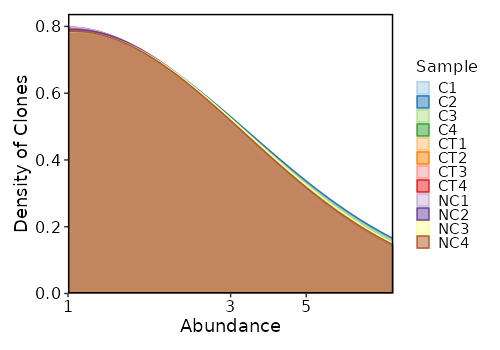{: width="80%"}
|
|
1868
|
+
|
|
1869
|
+
### CDR3 Length
|
|
1870
|
+
|
|
1871
|
+
```toml
|
|
1872
|
+
[ClonalStats.envs.cases."CDR3 Length"]
|
|
1873
|
+
viz_type = "length"
|
|
1874
|
+
```
|
|
1875
|
+
|
|
1876
|
+
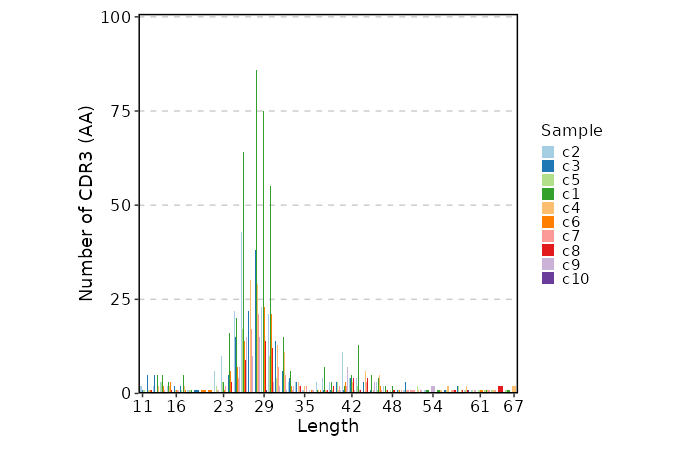{: width="80%"}
|
|
1877
|
+
|
|
1878
|
+
### CDR3 Length (Beta Chain)
|
|
1879
|
+
|
|
1880
|
+
```toml
|
|
1881
|
+
[ClonalStats.envs.cases."CDR3 Length (Beta Chain)"]
|
|
1882
|
+
viz_type = "length"
|
|
1883
|
+
chain = "TRB"
|
|
1884
|
+
```
|
|
1885
|
+
|
|
1886
|
+
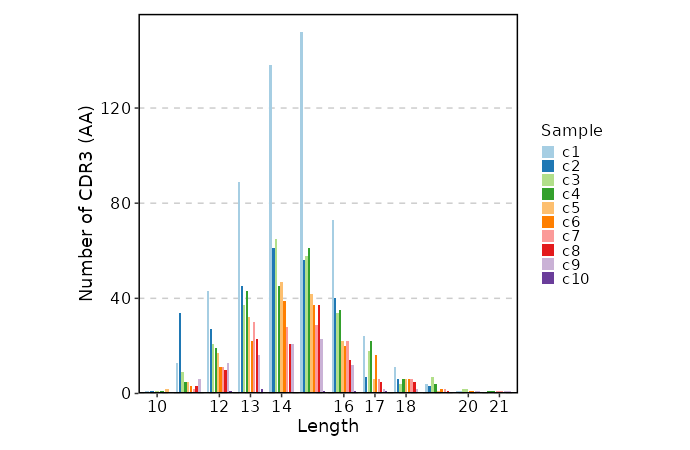{: width="80%"}
|
|
1887
|
+
|
|
1888
|
+
### Clonal Residency
|
|
1889
|
+
|
|
1890
|
+
```toml
|
|
1891
|
+
[ClonalStats.envs.cases."Clonal Residency"]
|
|
1892
|
+
viz_type = "residency"
|
|
1893
|
+
group_by = "Diagnosis"
|
|
1894
|
+
chain = "TRB"
|
|
1895
|
+
clone_call = "gene"
|
|
1896
|
+
groups = ["Colitis", "NoColitis"]
|
|
1897
|
+
```
|
|
1898
|
+
|
|
1899
|
+
{: width="80%"}
|
|
1900
|
+
|
|
1901
|
+
### Clonal Residency (UpSet Plot)
|
|
1902
|
+
|
|
1903
|
+
```toml
|
|
1904
|
+
[ClonalStats.envs.cases."Clonal Residency (UpSet Plot)"]
|
|
1905
|
+
viz_type = "residency"
|
|
1906
|
+
plot_type = "upset"
|
|
1907
|
+
group_by = "Diagnosis"
|
|
1908
|
+
chain = "TRB"
|
|
1909
|
+
clone_call = "gene"
|
|
1910
|
+
groups = ["Colitis", "NoColitis"]
|
|
1911
|
+
devpars = {width = 800}
|
|
1912
|
+
```
|
|
1913
|
+
|
|
1914
|
+
{: width="80%"}
|
|
1915
|
+
|
|
1916
|
+
### Clonal Statistics with Expanded Clones
|
|
1917
|
+
|
|
1918
|
+
```toml
|
|
1919
|
+
[ClonalStats.envs.cases."Clonal Statistics with Expanded Clones"]
|
|
1920
|
+
viz_type = "stat"
|
|
1921
|
+
plot_type = "pies"
|
|
1922
|
+
group_by = "Diagnosis"
|
|
1923
|
+
groups = ["Colitis", "NoColitis"]
|
|
1924
|
+
clones = {"Expanded Clones In Colitis" = "sel(Colitis > 2)", "Expanded Clones In NoColitis" = "sel(NoColitis > 2)"}
|
|
1925
|
+
subgroup_by = "seurat_clusters"
|
|
1926
|
+
pie_size = "sqrt"
|
|
1927
|
+
show_row_names = true
|
|
1928
|
+
show_column_names = true
|
|
1929
|
+
devpars = {width = 720}
|
|
1930
|
+
```
|
|
1931
|
+
|
|
1932
|
+
{: width="80%"}
|
|
1933
|
+
|
|
1934
|
+
### Hyperexpanded Clonal Dynamics
|
|
1935
|
+
|
|
1936
|
+
```toml
|
|
1937
|
+
[ClonalStats.envs.cases."Hyperexpanded Clonal Dynamics"]
|
|
1938
|
+
viz_type = "stat"
|
|
1939
|
+
plot_type = "sankey"
|
|
1940
|
+
group_by = "Diagnosis"
|
|
1941
|
+
chain = "TRB"
|
|
1942
|
+
groups = ["Colitis", "NoColitis"]
|
|
1943
|
+
clones = {"Hyper-Expanded Clones In Colitis" = "sel(Colitis > 5)", "Hyper-Expanded Clones In NoColitis" = "sel(NoColitis > 5)"}
|
|
1944
|
+
devpars = {width = 800}
|
|
1945
|
+
```
|
|
1946
|
+
|
|
1947
|
+
{: width="80%"}
|
|
1948
|
+
|
|
1949
|
+
### Clonal Composition
|
|
1950
|
+
|
|
1951
|
+
```toml
|
|
1952
|
+
[ClonalStats.envs.cases."Clonal Composition"]
|
|
1953
|
+
viz_type = "composition"
|
|
1954
|
+
x_text_angle = 45
|
|
1955
|
+
```
|
|
1956
|
+
|
|
1957
|
+
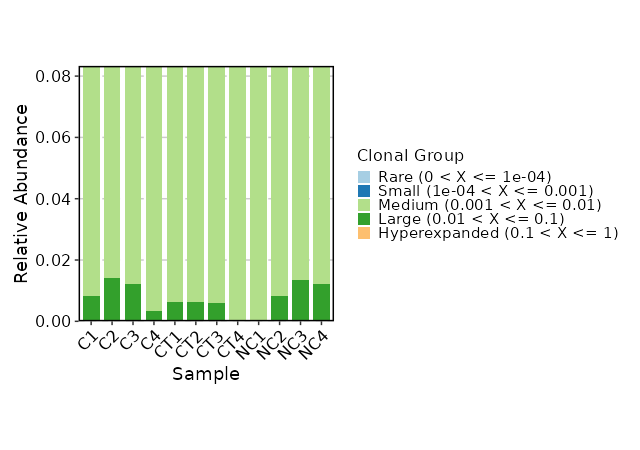{: width="80%"}
|
|
1958
|
+
|
|
1959
|
+
### Clonal Overlapping
|
|
1960
|
+
|
|
1961
|
+
```toml
|
|
1962
|
+
viz_type = "overlap"
|
|
1963
|
+
chain = "TRB"
|
|
1964
|
+
clone_call = "gene"
|
|
1965
|
+
```
|
|
1966
|
+
|
|
1967
|
+
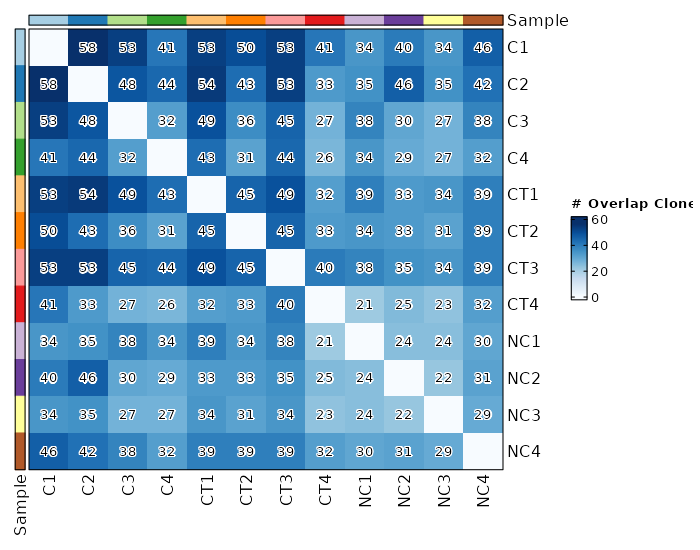{: width="80%"}
|
|
1968
|
+
|
|
1969
|
+
### Clonal Diversity
|
|
1970
|
+
|
|
1971
|
+
```toml
|
|
1972
|
+
[ClonalStats.envs.cases."Clonal Diversity"]
|
|
1973
|
+
# method = "shannon" # default
|
|
1974
|
+
viz_type = "diversity"
|
|
1975
|
+
x_text_angle = 45
|
|
1976
|
+
```
|
|
1977
|
+
|
|
1978
|
+
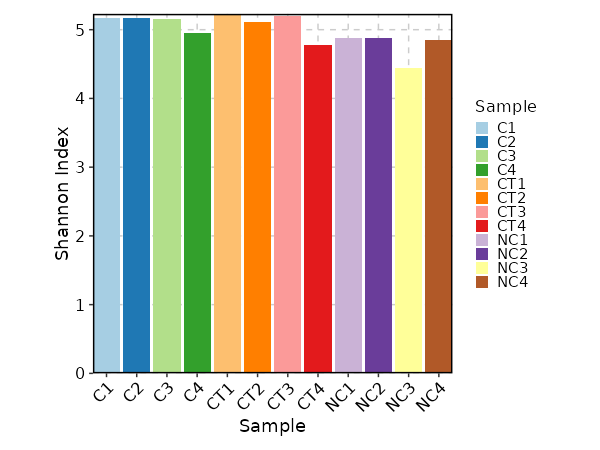{: width="80%"}
|
|
1979
|
+
|
|
1980
|
+
### Clonal Diversity (gini.coeff, by Diagnosis)
|
|
1981
|
+
|
|
1982
|
+
```toml
|
|
1983
|
+
[ClonalStats.envs.cases."Clonal Diversity (gini.coeff, by Diagnosis)"]
|
|
1984
|
+
method = "gini.coeff"
|
|
1985
|
+
viz_type = "diversity"
|
|
1986
|
+
plot_type = "box"
|
|
1987
|
+
group_by = "Diagnosis"
|
|
1988
|
+
comparisons = true
|
|
1989
|
+
devpars = {height = 600, width = 600}
|
|
1990
|
+
```
|
|
1991
|
+
|
|
1992
|
+
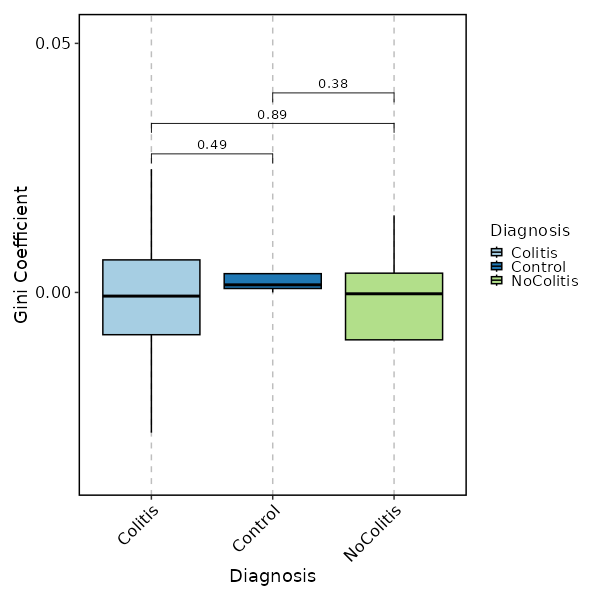{: width="80%"}
|
|
1993
|
+
|
|
1994
|
+
### Gene Usage Frequency
|
|
1995
|
+
|
|
1996
|
+
```toml
|
|
1997
|
+
[ClonalStats.envs.cases."Gene Usage Frequency"]
|
|
1998
|
+
viz_type = "geneusage"
|
|
1999
|
+
devpars = {width = 1200}
|
|
2000
|
+
```
|
|
2001
|
+
|
|
2002
|
+
{: width="80%"}
|
|
2003
|
+
|
|
2004
|
+
### Positional amino acid frequency
|
|
2005
|
+
|
|
2006
|
+
```toml
|
|
2007
|
+
[ClonalStats.envs.cases."Positional amino acid frequency"]
|
|
2008
|
+
viz_type = "positional"
|
|
2009
|
+
# method = "AA" # default
|
|
2010
|
+
devpars = {width = 1600}
|
|
2011
|
+
```
|
|
2012
|
+
|
|
2013
|
+
{: width="80%"}
|
|
2014
|
+
|
|
2015
|
+
### Positional shannon entropy
|
|
2016
|
+
|
|
2017
|
+
```toml
|
|
2018
|
+
[ClonalStats.envs.cases."Positional shannon entropy"]
|
|
2019
|
+
viz_type = "positional"
|
|
2020
|
+
method = "shannon"
|
|
2021
|
+
devpars = {width = 1200}
|
|
2022
|
+
```
|
|
2023
|
+
|
|
2024
|
+
{: width="80%"}
|
|
2025
|
+
|
|
2026
|
+
### 3-Mer Frequency
|
|
2027
|
+
|
|
2028
|
+
```toml
|
|
2029
|
+
[ClonalStats.envs.cases."3-Mer Frequency"]
|
|
2030
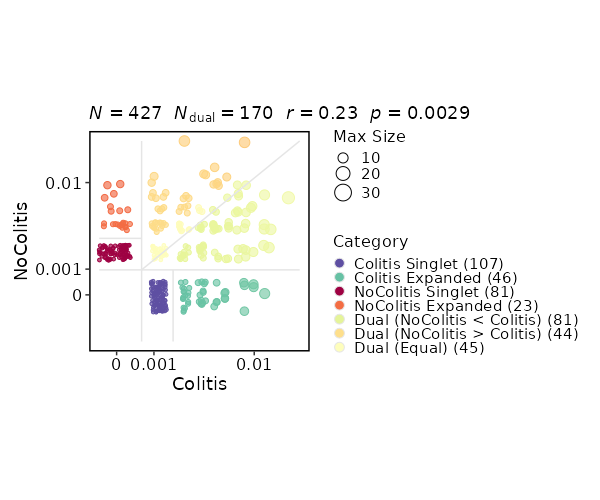
|
+
viz_type = "kmer"
|
|
2031
|
+
k = 3 # default is 3
|
|
2032
|
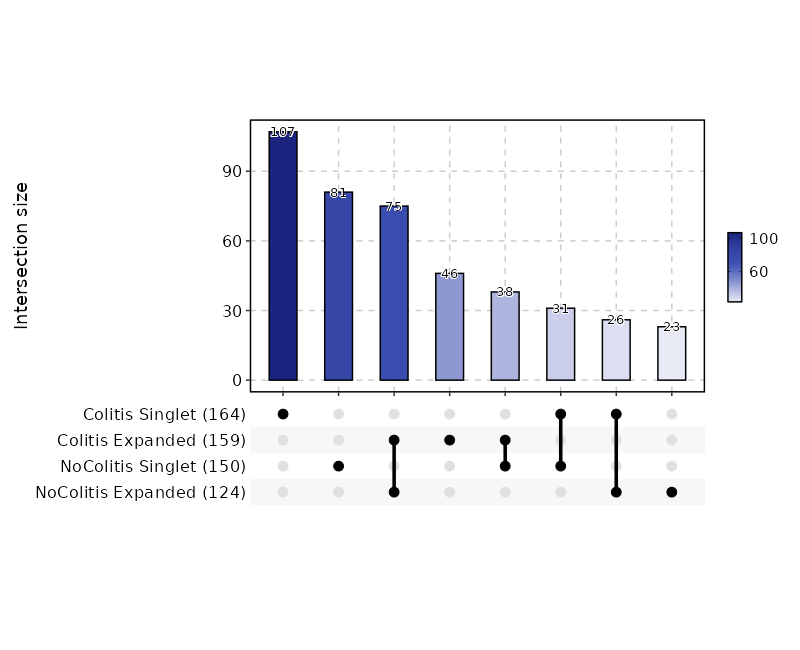
+
devpars = {width = 800}
|
|
2033
|
+
```
|
|
2034
|
+
|
|
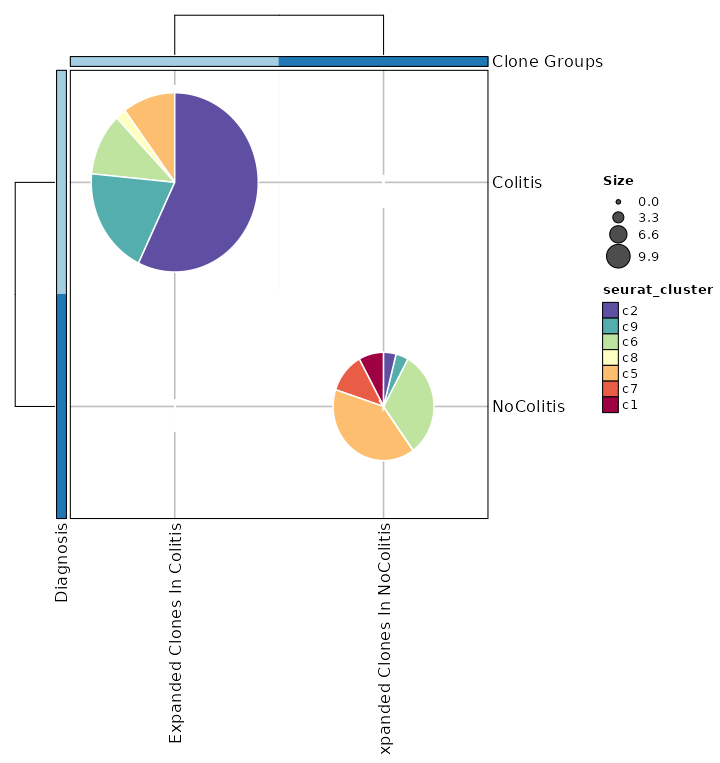
2035
|
+
{: width="80%"}
|
|
2036
|
+
|
|
2037
|
+
### Rarefaction Curve
|
|
2038
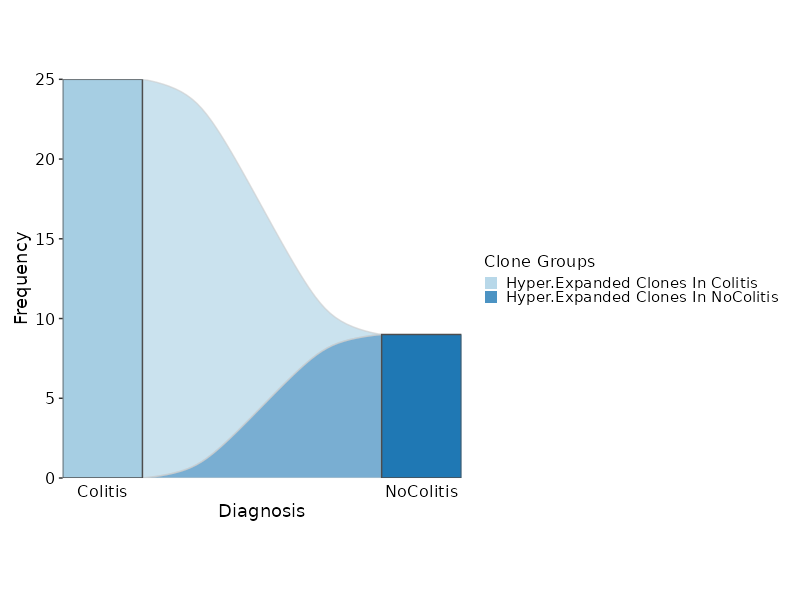
|
+
|
|
2039
|
+
```toml
|
|
2040
|
+
[ClonalStats.envs.cases."Rarefaction Curve"]
|
|
2041
|
+
viz_type = "rarefaction"
|
|
2042
|
+
```
|
|
2043
|
+
|
|
2044
|
+
{: width="80%"}
|
|
2045
|
+
|
|
1808
2046
|
Input:
|
|
1809
2047
|
screpfile: The `scRepertoire` object in RDS/qs format
|
|
1810
2048
|
|
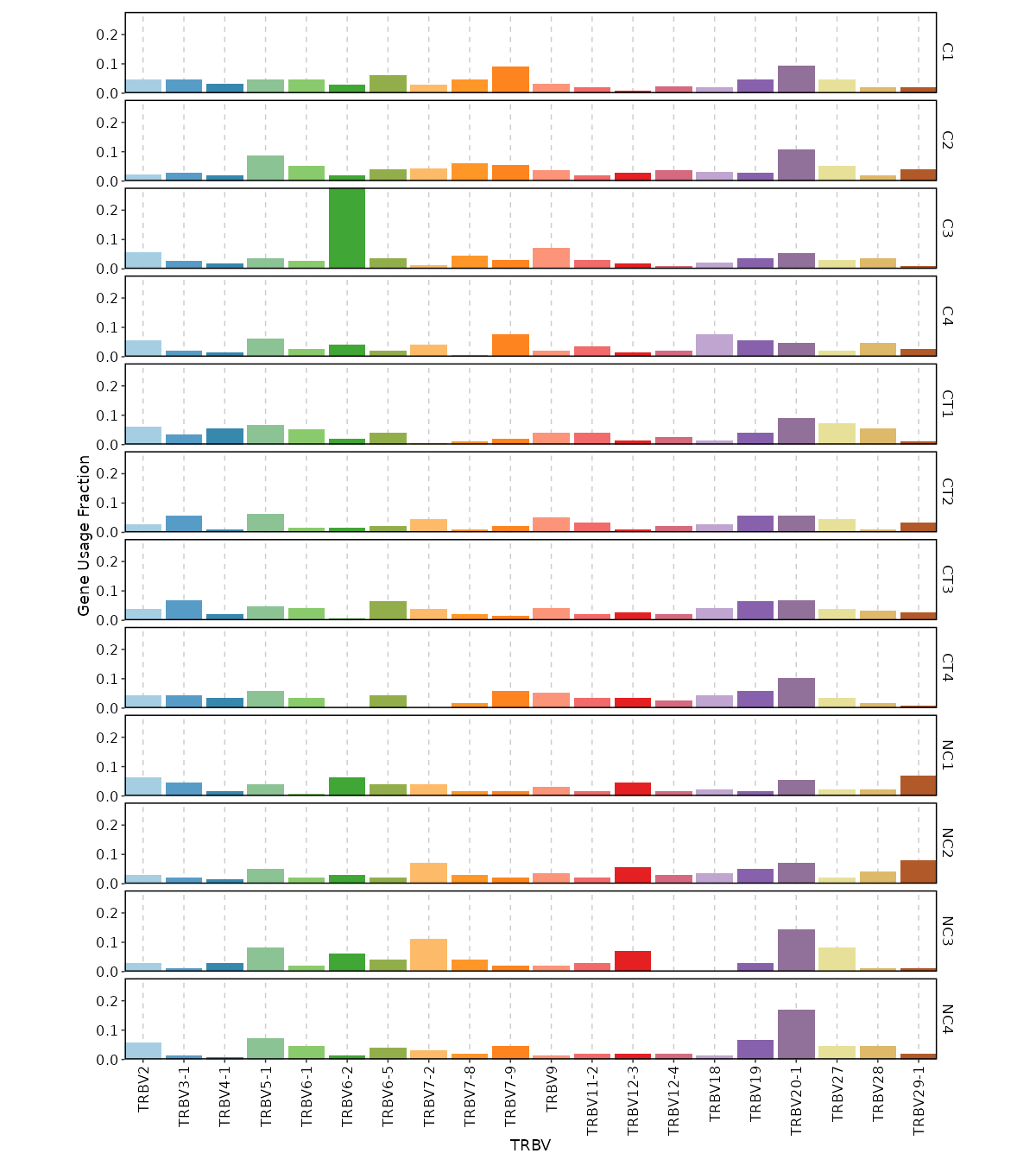
|
@@ -1822,7 +2060,7 @@ class ClonalStats(Proc):
|
|
|
1822
2060
|
- abundance: The abundance of the clones using [`ClonalAbundancePlot`](https://pwwang.github.io/scplotter/reference/ClonalAbundancePlot.html)
|
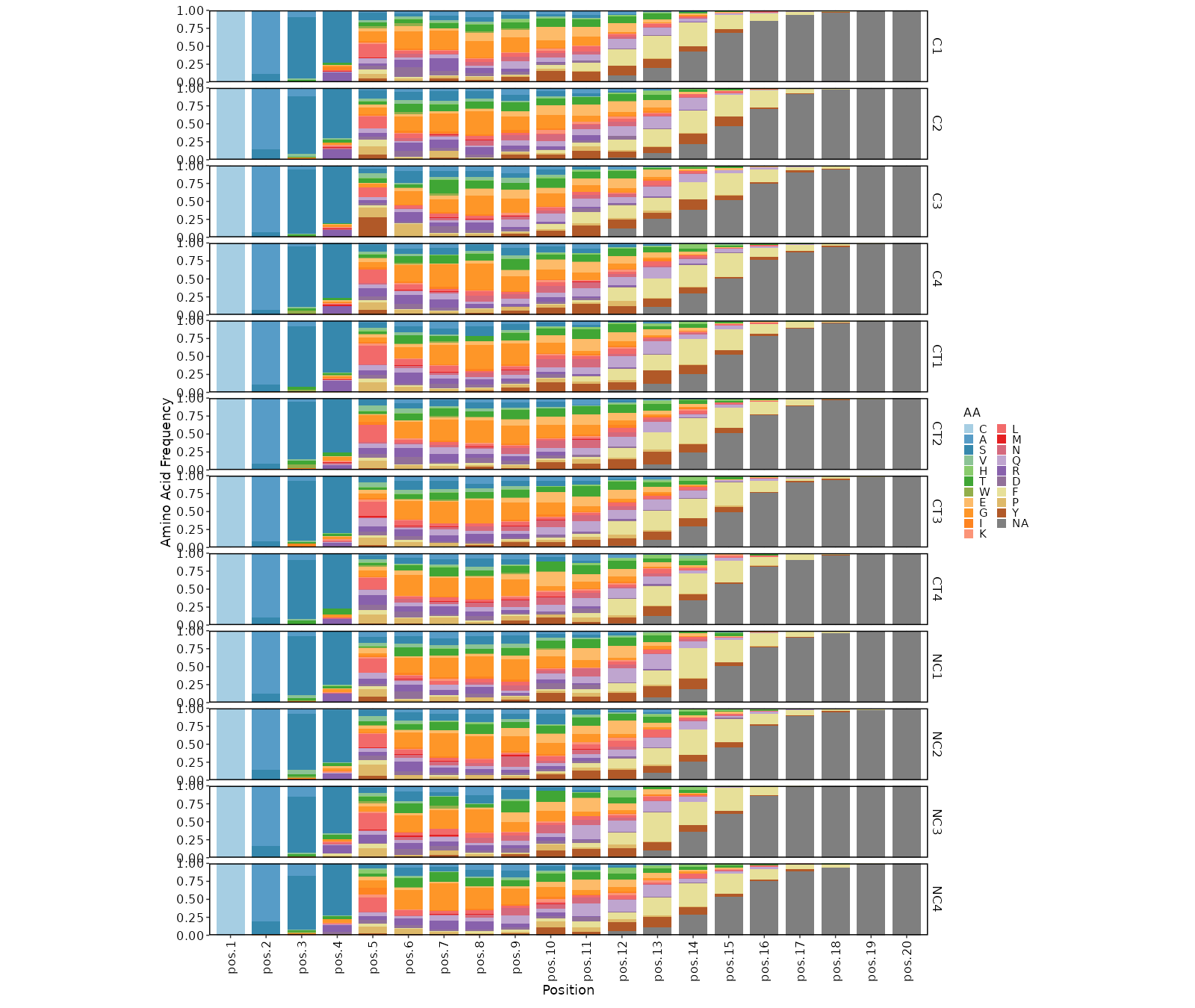
|
1823
2061
|
- length: The length of the CDR3 sequences using [`ClonalLengthPlot`](https://pwwang.github.io/scplotter/reference/ClonalLengthPlot.html)
|
|
1824
2062
|
- residency: The residency of the clones using [`ClonalResidencyPlot`](https://pwwang.github.io/scplotter/reference/ClonalResidencyPlot.html)
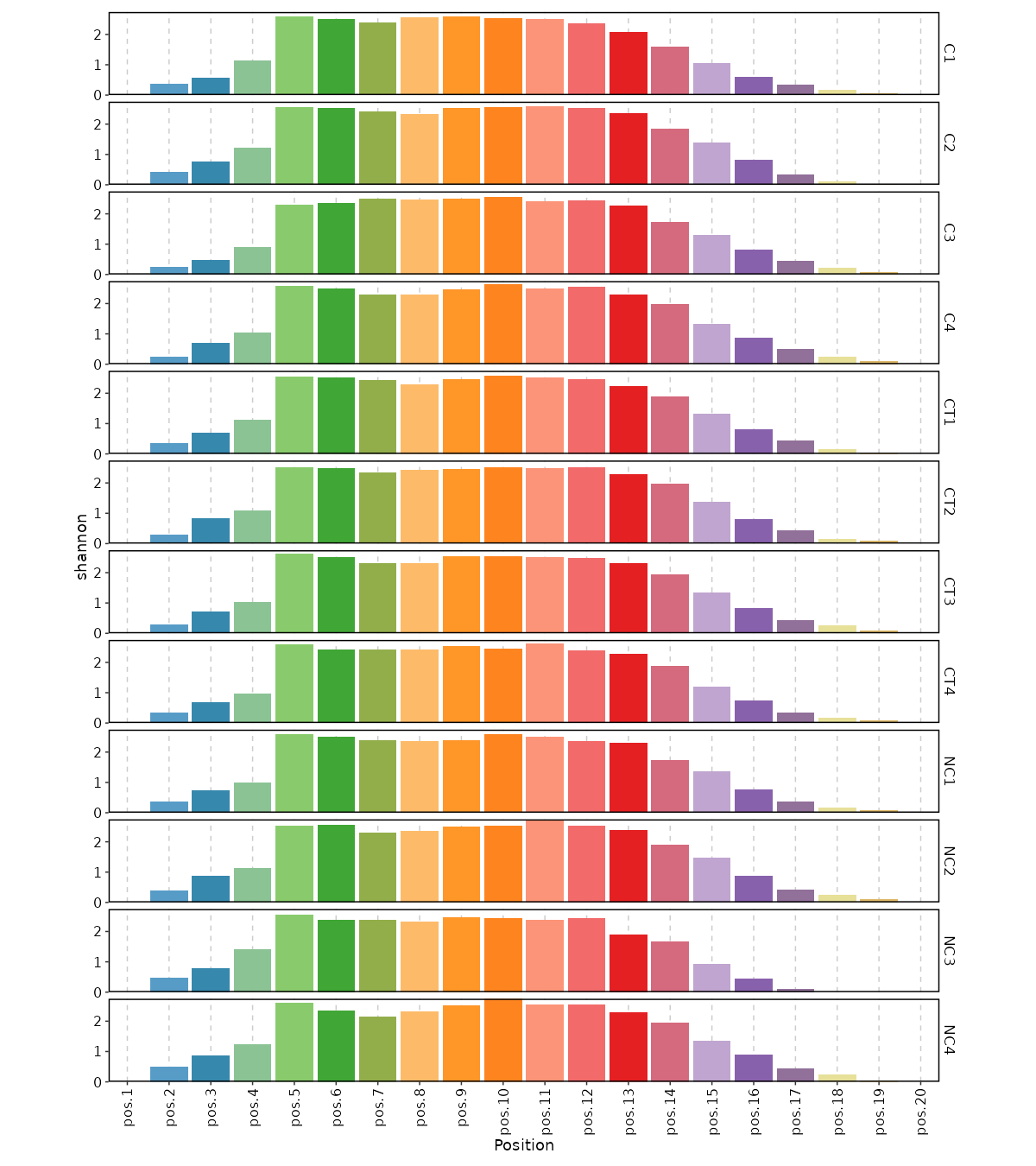
|
|
1825
|
-
-
|
|
2063
|
+
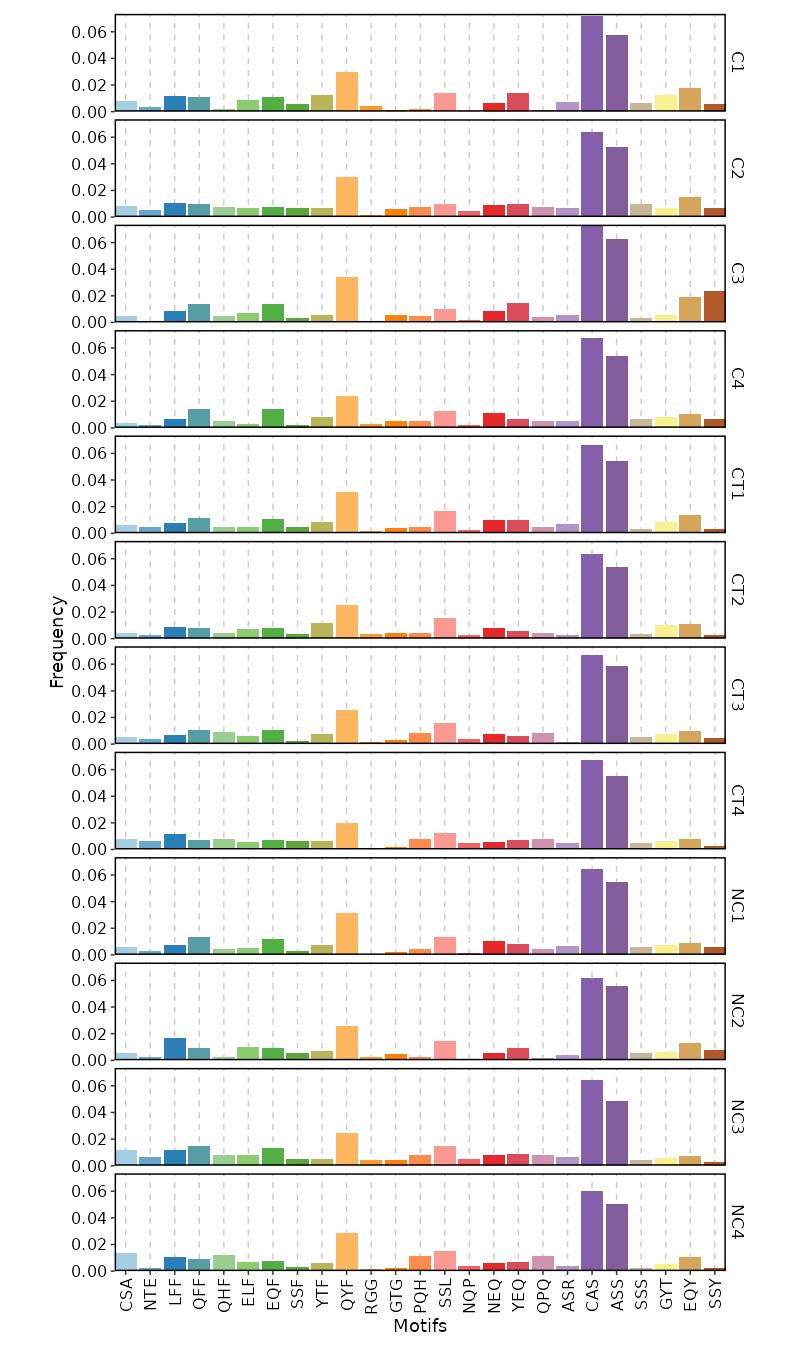
- stats: The stats of the clones using [`ClonalStatsPlot`](https://pwwang.github.io/scplotter/reference/ClonalStatsPlot.html)
|
|
1826
2064
|
- composition: The composition of the clones using [`ClonalCompositionPlot`](https://pwwang.github.io/scplotter/reference/ClonalCompositionPlot.html)
|
|
1827
2065
|
- overlap: The overlap of the clones using [`ClonalOverlapPlot`](https://pwwang.github.io/scplotter/reference/ClonalOverlapPlot.html)
|
|
1828
2066
|
- diversity: The diversity of the clones using [`ClonalDiversityPlot`](https://pwwang.github.io/scplotter/reference/ClonalDiversityPlot.html)
|
|
@@ -1842,6 +2080,7 @@ class ClonalStats(Proc):
|
|
|
1842
2080
|
save_code (flag): Whether to save the code used to generate the plots
|
|
1843
2081
|
Note that the data directly used to generate the plots will also be saved in an `rda` file.
|
|
1844
2082
|
Be careful if the data is large as it may take a lot of disk space.
|
|
2083
|
+
save_data (flag): Whether to save the data used to generate the plot.
|
|
1845
2084
|
descr: The description of the plot, used to show in the report.
|
|
1846
2085
|
<more>: The arguments for the plot function
|
|
1847
2086
|
See the documentation of the corresponding plot function for the details
|
|
@@ -1865,6 +2104,7 @@ class ClonalStats(Proc):
|
|
|
1865
2104
|
"devpars": {"width": None, "height": None, "res": 100},
|
|
1866
2105
|
"more_formats": [],
|
|
1867
2106
|
"save_code": False,
|
|
2107
|
+
"save_data": False,
|
|
1868
2108
|
"descr": None,
|
|
1869
2109
|
"cases": {
|
|
1870
2110
|
"Clonal Volume": {"viz_type": "volume"},
|
biopipen/ns/web.py
CHANGED
|
@@ -31,8 +31,13 @@ class Download(Proc):
|
|
|
31
31
|
"""
|
|
32
32
|
input = "url"
|
|
33
33
|
output = (
|
|
34
|
+
# Need to replace http:// and https:// to avoid cloudpathlib.AnyPath to get
|
|
35
|
+
# the basename for something like "https://example.com/data/?file=datafile.txt"
|
|
36
|
+
# as data, but "?file=datafile.txt"
|
|
34
37
|
"outfile:file:"
|
|
35
38
|
"""{{in.url
|
|
39
|
+
| replace: 'http://', ''
|
|
40
|
+
| replace: 'https://', ''
|
|
36
41
|
| basename
|
|
37
42
|
| url_decode
|
|
38
43
|
| slugify: separator='.', lowercase=False, regex_pattern='[^-a-zA-Z0-9_]+'
|
|
@@ -50,6 +50,15 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
50
50
|
</ListItem>
|
|
51
51
|
</UnorderedList>
|
|
52
52
|
|
|
53
|
+
<style>
|
|
54
|
+
.listitem {
|
|
55
|
+
font-size: large;
|
|
56
|
+
font-weight: bold;
|
|
57
|
+
margin: 1rem 0 0.5rem 0;
|
|
58
|
+
display: inline-block;
|
|
59
|
+
}
|
|
60
|
+
</style>
|
|
61
|
+
|
|
53
62
|
{%- macro report_job(job, h=1) -%}
|
|
54
63
|
{{ job | render_job: h=h }}
|
|
55
64
|
{%- endmacro -%}
|
|
@@ -59,12 +68,3 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
59
68
|
{%- endmacro -%}
|
|
60
69
|
|
|
61
70
|
{{ report_jobs(jobs, head_job, report_job) }}
|
|
62
|
-
|
|
63
|
-
<style>
|
|
64
|
-
.listitem {
|
|
65
|
-
font-size: large;
|
|
66
|
-
font-weight: bold;
|
|
67
|
-
margin: 1rem 0 0.5rem 0;
|
|
68
|
-
display: inline-block;
|
|
69
|
-
}
|
|
70
|
-
</style>
|
|
@@ -82,6 +82,15 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
82
82
|
</ListItem>
|
|
83
83
|
</UnorderedList>
|
|
84
84
|
|
|
85
|
+
<style>
|
|
86
|
+
.listitem {
|
|
87
|
+
font-size: large;
|
|
88
|
+
font-weight: bold;
|
|
89
|
+
margin: 1rem 0 0.5rem 0;
|
|
90
|
+
display: inline-block;
|
|
91
|
+
}
|
|
92
|
+
</style>
|
|
93
|
+
|
|
85
94
|
{%- macro report_job(job, h=1) -%}
|
|
86
95
|
{{ job | render_job: h=h }}
|
|
87
96
|
{%- endmacro -%}
|
|
@@ -92,11 +101,3 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
92
101
|
|
|
93
102
|
{{ report_jobs(jobs, head_job, report_job) }}
|
|
94
103
|
|
|
95
|
-
<style>
|
|
96
|
-
.listitem {
|
|
97
|
-
font-size: large;
|
|
98
|
-
font-weight: bold;
|
|
99
|
-
margin: 1rem 0 0.5rem 0;
|
|
100
|
-
display: inline-block;
|
|
101
|
-
}
|
|
102
|
-
</style>
|
|
@@ -61,6 +61,15 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
61
61
|
</ListItem>
|
|
62
62
|
</UnorderedList>
|
|
63
63
|
|
|
64
|
+
<style>
|
|
65
|
+
.listitem {
|
|
66
|
+
font-size: large;
|
|
67
|
+
font-weight: bold;
|
|
68
|
+
margin: 1rem 0 0.5rem 0;
|
|
69
|
+
display: inline-block;
|
|
70
|
+
}
|
|
71
|
+
</style>
|
|
72
|
+
|
|
64
73
|
{%- macro report_job(job, h=1) -%}
|
|
65
74
|
{{ job | render_job: h=h }}
|
|
66
75
|
{%- endmacro -%}
|
|
@@ -70,12 +79,3 @@ The cells are grouped at 2 dimensions: `subset_by`, usually the clinic groups th
|
|
|
70
79
|
{%- endmacro -%}
|
|
71
80
|
|
|
72
81
|
{{ report_jobs(jobs, head_job, report_job) }}
|
|
73
|
-
|
|
74
|
-
<style>
|
|
75
|
-
.listitem {
|
|
76
|
-
font-size: large;
|
|
77
|
-
font-weight: bold;
|
|
78
|
-
margin: 1rem 0 0.5rem 0;
|
|
79
|
-
display: inline-block;
|
|
80
|
-
}
|
|
81
|
-
</style>
|
|
@@ -1,12 +1,13 @@
|
|
|
1
|
-
import
|
|
1
|
+
from contextlib import suppress
|
|
2
|
+
import hashlib
|
|
3
|
+
import shutil
|
|
2
4
|
import re
|
|
3
|
-
import os.path
|
|
4
5
|
from pathlib import Path, PosixPath # noqa: F401
|
|
5
6
|
from biopipen.utils.misc import run_command
|
|
6
7
|
|
|
7
8
|
fastqs: list[Path] = {{in.fastqs | each: as_path}} # pyright: ignore # noqa
|
|
8
|
-
outdir:
|
|
9
|
-
id = {{out.outdir | basename | quote}} # pyright: ignore
|
|
9
|
+
outdir: Path = Path({{out.outdir | quote}}) # pyright: ignore
|
|
10
|
+
id: str = {{out.outdir | basename | quote}} # pyright: ignore
|
|
10
11
|
|
|
11
12
|
cellranger = {{envs.cellranger | quote}} # pyright: ignore
|
|
12
13
|
tmpdir = Path({{envs.tmpdir | quote}}) # pyright: ignore
|
|
@@ -14,12 +15,18 @@ ref: str = {{envs.ref | quote}} # pyright: ignore
|
|
|
14
15
|
ncores = {{envs.ncores | int}} # pyright: ignore
|
|
15
16
|
include_introns = {{envs.include_introns | repr}} # pyright: ignore
|
|
16
17
|
create_bam = {{envs.create_bam | repr}} # pyright: ignore
|
|
18
|
+
outdir_is_mounted: bool = {{envs.outdir_is_mounted | repr}} # pyright: ignore
|
|
19
|
+
copy_outs_only: bool = {{envs.copy_outs_only | repr}} # pyright: ignore
|
|
17
20
|
|
|
21
|
+
ref: Path = Path(ref).resolve() # pyright: ignore
|
|
22
|
+
if not ref.exists():
|
|
23
|
+
raise FileNotFoundError(f"Reference path does not exist: {ref}")
|
|
18
24
|
include_introns = str(include_introns).lower()
|
|
19
25
|
create_bam = str(create_bam).lower()
|
|
20
26
|
|
|
21
27
|
# create a temporary unique directory to store the soft-linked fastq files
|
|
22
|
-
|
|
28
|
+
uid = hashlib.md5(str(fastqs).encode()).hexdigest()[:8]
|
|
29
|
+
fastqdir = tmpdir / f"cellranger_count_{uid}"
|
|
23
30
|
fastqdir.mkdir(parents=True, exist_ok=True)
|
|
24
31
|
if len(fastqs) == 1 and fastqs[0].is_dir():
|
|
25
32
|
fastqs = list(fastqs[0].glob("*.fastq.gz"))
|
|
@@ -39,7 +46,7 @@ for fastq in fastqs:
|
|
|
39
46
|
|
|
40
47
|
linked.symlink_to(fastq)
|
|
41
48
|
|
|
42
|
-
other_args = {{envs | dict_to_cli_args: dashify=True, exclude=['no_bam', 'create_bam', 'include_introns', 'cellranger', 'transcriptome', 'ref', 'tmpdir', 'id', 'ncores']}} # pyright: ignore
|
|
49
|
+
other_args = {{envs | dict_to_cli_args: dashify=True, exclude=['no_bam', 'create_bam', 'include_introns', 'cellranger', 'transcriptome', 'ref', 'tmpdir', 'id', 'ncores', 'outdir_is_mounted', 'copy_outs_only']}} # pyright: ignore
|
|
43
50
|
|
|
44
51
|
command = [
|
|
45
52
|
cellranger,
|
|
@@ -49,7 +56,7 @@ command = [
|
|
|
49
56
|
"--fastqs",
|
|
50
57
|
fastqdir,
|
|
51
58
|
"--transcriptome",
|
|
52
|
-
|
|
59
|
+
str(ref),
|
|
53
60
|
"--localcores",
|
|
54
61
|
ncores,
|
|
55
62
|
"--disable-ui",
|
|
@@ -62,18 +69,29 @@ command = [
|
|
|
62
69
|
# cellranger cellranger-7.2.0
|
|
63
70
|
version: str = run_command([cellranger, "--version"], stdout = "RETURN") # type: ignore
|
|
64
71
|
version = version.replace("cellranger", "").replace("-", "").strip() # type: ignore
|
|
72
|
+
print(f"# Detected cellranger version: {version}")
|
|
65
73
|
version: list[int] = list(map(int, version.split("."))) # type: ignore
|
|
66
74
|
if version[0] >= 8:
|
|
67
75
|
command += ["--create-bam", create_bam]
|
|
68
76
|
elif create_bam != "true":
|
|
69
77
|
command += ["--no-bam"]
|
|
70
78
|
|
|
71
|
-
|
|
79
|
+
if outdir_is_mounted:
|
|
80
|
+
print("# Using mounted outdir, redirecting cellranger output to a local tmpdir")
|
|
81
|
+
local_outdir = tmpdir / f"{outdir.name}-{uid}" / id
|
|
82
|
+
if local_outdir.parent.exists():
|
|
83
|
+
shutil.rmtree(local_outdir.parent)
|
|
84
|
+
local_outdir.parent.mkdir(parents=True, exist_ok=True)
|
|
85
|
+
odir = local_outdir
|
|
86
|
+
else:
|
|
87
|
+
odir = outdir
|
|
72
88
|
|
|
73
|
-
|
|
89
|
+
run_command(command, fg=True, cwd=str(odir.parent))
|
|
90
|
+
|
|
91
|
+
web_summary_html = odir / "outs" / "web_summary.html"
|
|
74
92
|
if not web_summary_html.exists():
|
|
75
93
|
raise RuntimeError(
|
|
76
|
-
f"web_summary.html does not exist in {
|
|
94
|
+
f"web_summary.html does not exist in {odir}/outs. "
|
|
77
95
|
"cellranger count failed."
|
|
78
96
|
)
|
|
79
97
|
|
|
@@ -81,7 +99,7 @@ if not web_summary_html.exists():
|
|
|
81
99
|
# to void vscode live server breaking the page by injecting some code
|
|
82
100
|
print("# Modify web_summary.html to move javascript to a separate file")
|
|
83
101
|
try:
|
|
84
|
-
web_summary_js =
|
|
102
|
+
web_summary_js = odir / "outs" / "web_summary.js"
|
|
85
103
|
web_summary_content = web_summary_html.read_text()
|
|
86
104
|
regex = re.compile(r"<script>(.+)</script>", re.DOTALL)
|
|
87
105
|
web_summary_html.write_text(regex.sub(
|
|
@@ -92,3 +110,29 @@ try:
|
|
|
92
110
|
except Exception as e:
|
|
93
111
|
print(f"Error modifying web_summary.html: {e}")
|
|
94
112
|
raise e
|
|
113
|
+
|
|
114
|
+
# If using local tmpdir for output, move results to the final outdir
|
|
115
|
+
if outdir_is_mounted:
|
|
116
|
+
print("# Copy results back to outdir")
|
|
117
|
+
if outdir.exists():
|
|
118
|
+
shutil.rmtree(outdir)
|
|
119
|
+
|
|
120
|
+
if copy_outs_only:
|
|
121
|
+
outdir.mkdir(parents=True, exist_ok=True)
|
|
122
|
+
with suppress(Exception):
|
|
123
|
+
# Some files may be failed to copy due to permission issues
|
|
124
|
+
# But the contents are actually copied
|
|
125
|
+
shutil.copytree(odir / "outs", outdir / "outs")
|
|
126
|
+
else:
|
|
127
|
+
with suppress(Exception):
|
|
128
|
+
shutil.copytree(local_outdir, outdir) # type: ignore
|
|
129
|
+
|
|
130
|
+
# Make sure essential files exist
|
|
131
|
+
web_summary_html = outdir / "outs" / "web_summary.html"
|
|
132
|
+
web_summary_js = outdir / "outs" / "web_summary.js"
|
|
133
|
+
for f in [web_summary_html, web_summary_js]:
|
|
134
|
+
if not f.exists():
|
|
135
|
+
raise RuntimeError(
|
|
136
|
+
f"{f} does not exist in {outdir}/outs. "
|
|
137
|
+
"Copying results back from tmpdir failed."
|
|
138
|
+
)
|