pylocuszoom 1.1.2__py3-none-any.whl → 1.3.1__py3-none-any.whl
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- pylocuszoom/__init__.py +20 -2
- pylocuszoom/backends/base.py +94 -2
- pylocuszoom/backends/bokeh_backend.py +160 -6
- pylocuszoom/backends/matplotlib_backend.py +142 -2
- pylocuszoom/backends/plotly_backend.py +101 -1
- pylocuszoom/coloc.py +82 -0
- pylocuszoom/coloc_plotter.py +390 -0
- pylocuszoom/colors.py +26 -0
- pylocuszoom/config.py +61 -0
- pylocuszoom/finemapping.py +111 -3
- pylocuszoom/labels.py +41 -16
- pylocuszoom/ld.py +239 -0
- pylocuszoom/ld_heatmap_plotter.py +252 -0
- pylocuszoom/miami_plotter.py +490 -0
- pylocuszoom/plotter.py +483 -342
- pylocuszoom/recombination.py +39 -0
- {pylocuszoom-1.1.2.dist-info → pylocuszoom-1.3.1.dist-info}/METADATA +183 -31
- {pylocuszoom-1.1.2.dist-info → pylocuszoom-1.3.1.dist-info}/RECORD +20 -16
- pylocuszoom-1.3.1.dist-info/licenses/LICENSE.md +595 -0
- pylocuszoom-1.1.2.dist-info/licenses/LICENSE.md +0 -17
- {pylocuszoom-1.1.2.dist-info → pylocuszoom-1.3.1.dist-info}/WHEEL +0 -0
pylocuszoom/recombination.py
CHANGED
|
@@ -395,6 +395,45 @@ def get_recombination_rate_for_region(
|
|
|
395
395
|
return region_df[["pos", "rate"]]
|
|
396
396
|
|
|
397
397
|
|
|
398
|
+
def ensure_recomb_maps(
|
|
399
|
+
species: str = "canine",
|
|
400
|
+
data_dir: Optional[str] = None,
|
|
401
|
+
) -> Optional[Path]:
|
|
402
|
+
"""Ensure recombination maps are available, downloading if needed.
|
|
403
|
+
|
|
404
|
+
Args:
|
|
405
|
+
species: Species name ('canine', 'feline', etc.).
|
|
406
|
+
data_dir: Directory for recombination maps. Uses default if None.
|
|
407
|
+
|
|
408
|
+
Returns:
|
|
409
|
+
Path to recombination maps directory, or None if species not supported
|
|
410
|
+
or download fails.
|
|
411
|
+
"""
|
|
412
|
+
if species != "canine":
|
|
413
|
+
logger.debug(f"No built-in recombination maps for species: {species}")
|
|
414
|
+
return None
|
|
415
|
+
|
|
416
|
+
if data_dir is not None:
|
|
417
|
+
output_path = Path(data_dir)
|
|
418
|
+
else:
|
|
419
|
+
output_path = get_default_data_dir()
|
|
420
|
+
|
|
421
|
+
# Check if maps already exist
|
|
422
|
+
if output_path.exists():
|
|
423
|
+
existing_files = list(output_path.glob("chr*_recomb.tsv"))
|
|
424
|
+
if len(existing_files) >= 39: # 38 autosomes + X
|
|
425
|
+
logger.debug(f"Recombination maps already exist at {output_path}")
|
|
426
|
+
return output_path
|
|
427
|
+
|
|
428
|
+
# Download maps with error handling
|
|
429
|
+
logger.info("Downloading canine recombination maps...")
|
|
430
|
+
try:
|
|
431
|
+
return download_canine_recombination_maps(output_dir=str(output_path))
|
|
432
|
+
except Exception as e:
|
|
433
|
+
logger.warning(f"Could not download recombination maps: {e}")
|
|
434
|
+
return None
|
|
435
|
+
|
|
436
|
+
|
|
398
437
|
def add_recombination_overlay(
|
|
399
438
|
ax: Axes,
|
|
400
439
|
recomb_df: pd.DataFrame,
|
|
@@ -1,6 +1,6 @@
|
|
|
1
1
|
Metadata-Version: 2.4
|
|
2
2
|
Name: pylocuszoom
|
|
3
|
-
Version: 1.1
|
|
3
|
+
Version: 1.3.1
|
|
4
4
|
Summary: Publication-ready regional association plots with LD coloring, gene tracks, and recombination overlays
|
|
5
5
|
Project-URL: Homepage, https://github.com/michael-denyer/pylocuszoom
|
|
6
6
|
Project-URL: Documentation, https://github.com/michael-denyer/pylocuszoom#readme
|
|
@@ -35,6 +35,7 @@ Requires-Dist: tqdm>=4.60.0
|
|
|
35
35
|
Provides-Extra: all
|
|
36
36
|
Requires-Dist: pyspark>=3.0.0; extra == 'all'
|
|
37
37
|
Provides-Extra: dev
|
|
38
|
+
Requires-Dist: hypothesis>=6.0.0; extra == 'dev'
|
|
38
39
|
Requires-Dist: pytest-cov>=4.0.0; extra == 'dev'
|
|
39
40
|
Requires-Dist: pytest-randomly>=3.0.0; extra == 'dev'
|
|
40
41
|
Requires-Dist: pytest-xdist>=3.0.0; extra == 'dev'
|
|
@@ -71,20 +72,23 @@ Inspired by [LocusZoom](http://locuszoom.org/) and [locuszoomr](https://github.c
|
|
|
71
72
|
- **SNP labels (matplotlib)**: Automatic labeling of top SNPs by p-value (RS IDs)
|
|
72
73
|
- **Hover tooltips (Plotly and Bokeh)**: Detailed SNP data on hover
|
|
73
74
|
|
|
74
|
-

|
|
75
|
+
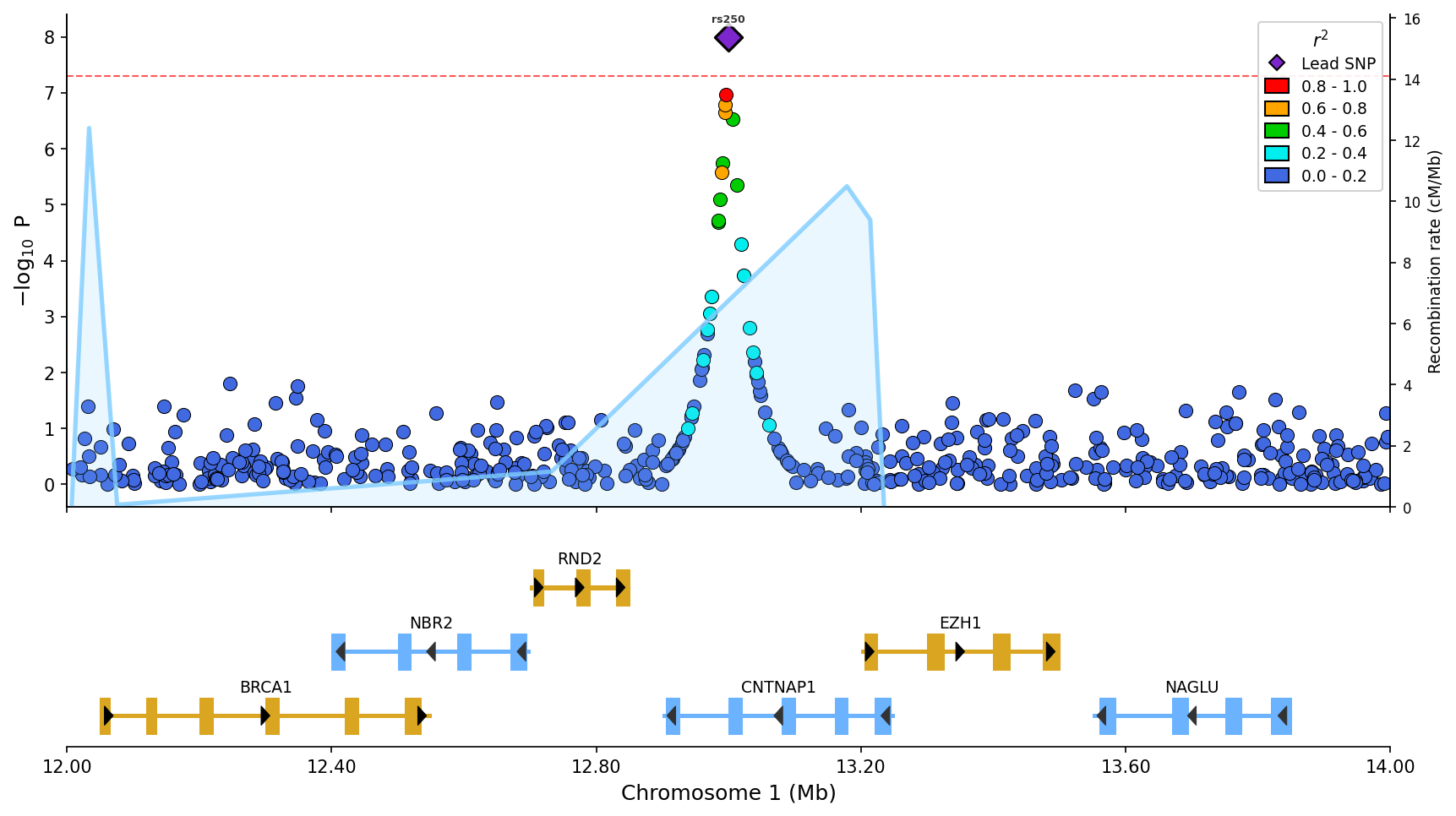
|
|
75
76
|
*Regional association plot with LD coloring, gene/exon track, recombination rate overlay (blue line), and top SNP labels.*
|
|
76
77
|
|
|
77
78
|
2. **Stacked plots**: Compare multiple GWAS/phenotypes vertically
|
|
78
|
-
3. **
|
|
79
|
-
4. **
|
|
80
|
-
5. **
|
|
81
|
-
6. **
|
|
82
|
-
7. **
|
|
83
|
-
8. **
|
|
84
|
-
9. **
|
|
85
|
-
10. **
|
|
86
|
-
11. **
|
|
87
|
-
12. **
|
|
79
|
+
3. **Miami plots**: Mirrored Manhattan plots for comparing two GWAS datasets (discovery vs replication)
|
|
80
|
+
4. **Manhattan plots**: Genome-wide association visualization with chromosome coloring
|
|
81
|
+
5. **QQ plots**: Quantile-quantile plots with confidence bands and genomic inflation factor
|
|
82
|
+
6. **eQTL plot**: Expression QTL data aligned with association plots and gene tracks
|
|
83
|
+
7. **Fine-mapping plots**: Visualize SuSiE credible sets with posterior inclusion probabilities
|
|
84
|
+
8. **PheWAS plots**: Phenome-wide association study visualization across multiple phenotypes
|
|
85
|
+
9. **Forest plots**: Meta-analysis effect size visualization with confidence intervals
|
|
86
|
+
10. **LD heatmaps**: Triangular heatmaps showing pairwise LD patterns, standalone or integrated below regional plots
|
|
87
|
+
11. **Colocalization plots**: GWAS-eQTL scatter plots with LD coloring, correlation statistics, and effect direction visualization
|
|
88
|
+
12. **Multiple backends**: matplotlib (publication-ready), plotly (interactive), bokeh (dashboard integration)
|
|
89
|
+
12. **Pandas and PySpark support**: Works with both Pandas and PySpark DataFrames for large-scale genomics data
|
|
90
|
+
13. **Convenience data file loaders**: Load and validate common GWAS, eQTL and fine-mapping file formats
|
|
91
|
+
14. **Automatic gene annotations**: Fetch gene/exon data from Ensembl REST API with caching (human, mouse, rat, canine, feline, and any Ensembl species)
|
|
88
92
|
|
|
89
93
|
## Installation
|
|
90
94
|
|
|
@@ -254,7 +258,7 @@ fig = plotter.plot_stacked(
|
|
|
254
258
|
)
|
|
255
259
|
```
|
|
256
260
|
|
|
257
|
-

|
|
261
|
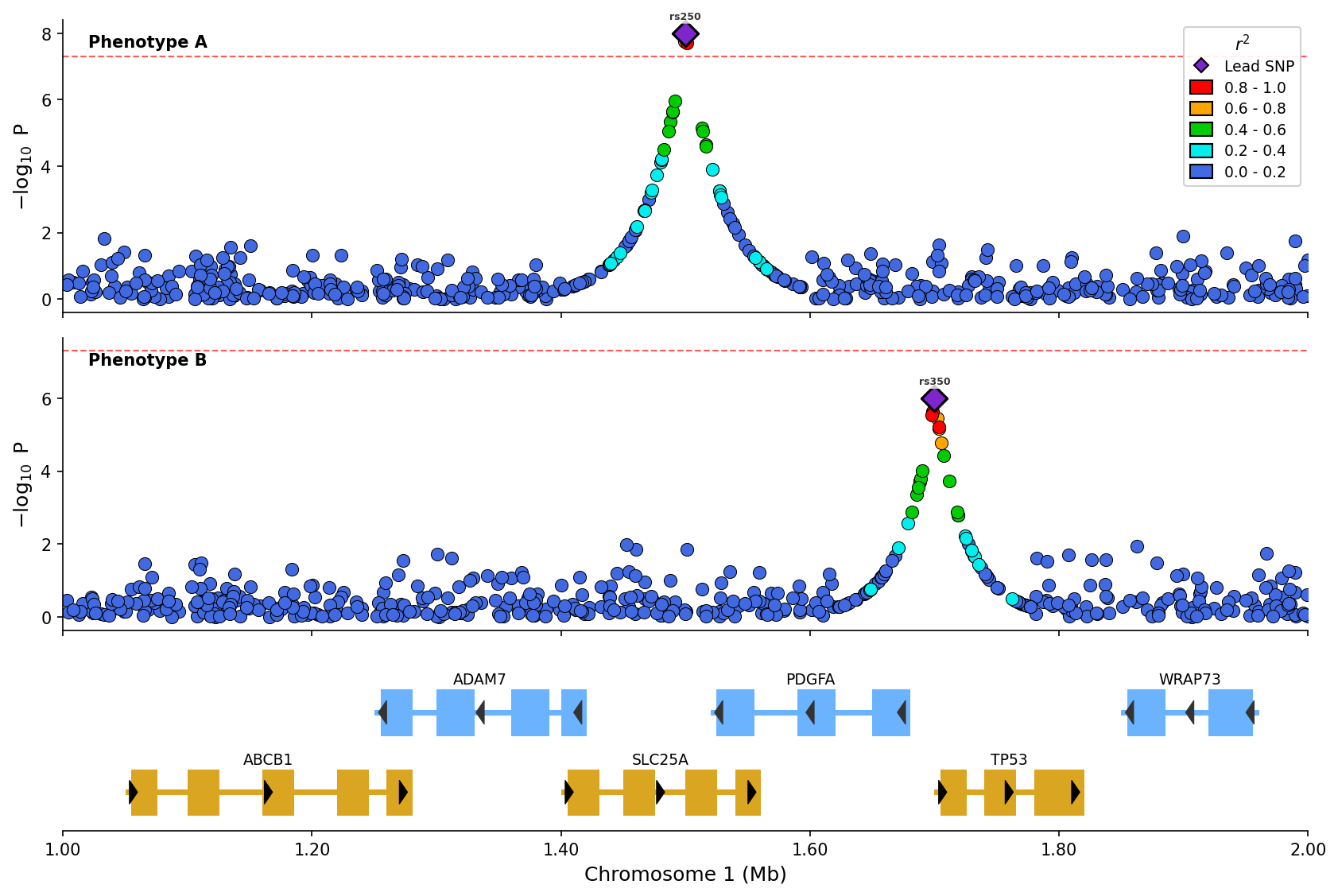
+
|
|
258
262
|
*Stacked plot comparing two phenotypes with LD coloring and shared gene track.*
|
|
259
263
|
|
|
260
264
|
## eQTL Overlay
|
|
@@ -283,7 +287,7 @@ fig = plotter.plot_stacked(
|
|
|
283
287
|
)
|
|
284
288
|
```
|
|
285
289
|
|
|
286
|
-

|
|
290
|
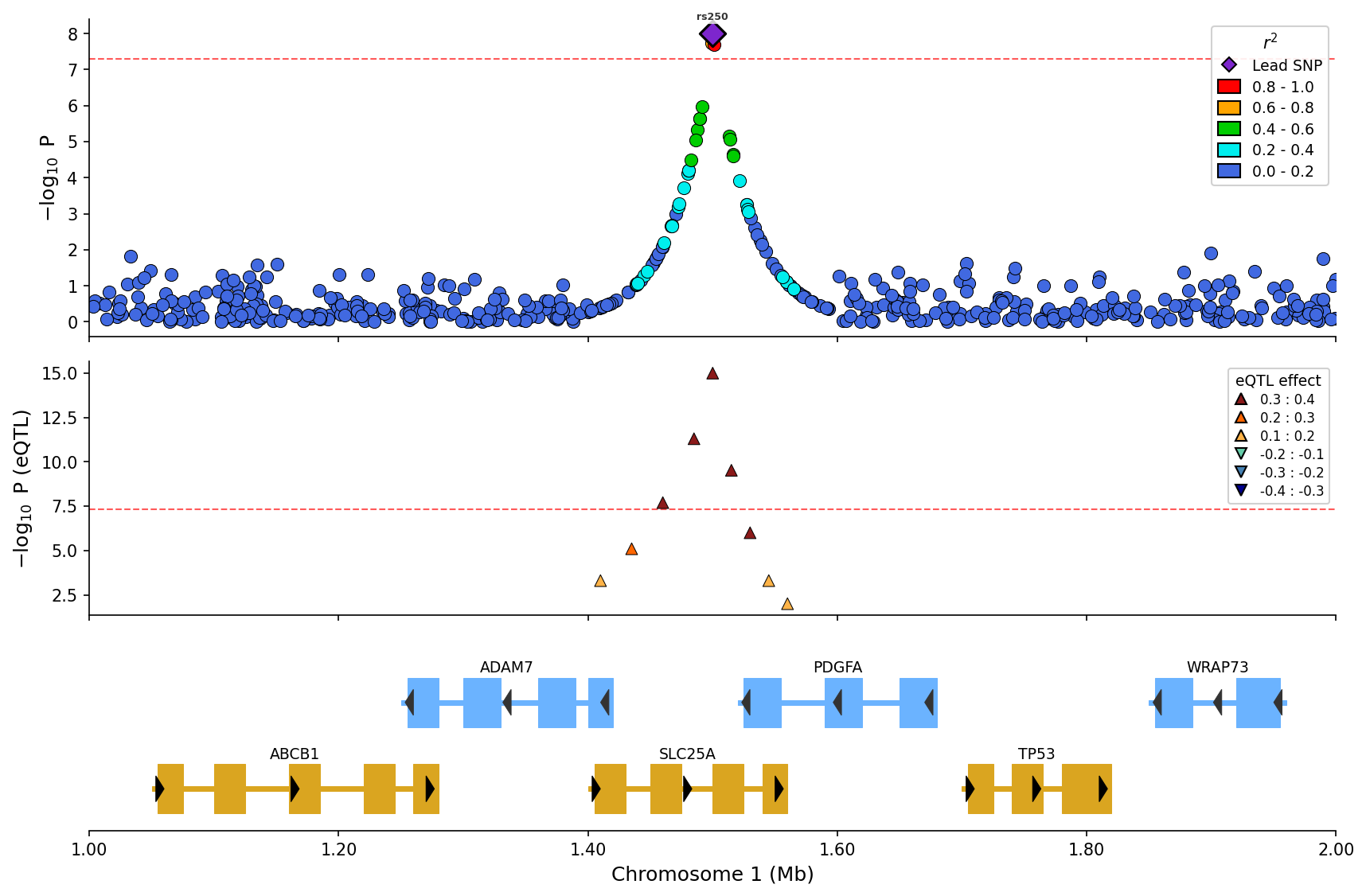
+
|
|
287
291
|
*eQTL overlay with effect direction (up/down triangles) and magnitude binning.*
|
|
288
292
|
|
|
289
293
|
## Fine-mapping Visualization
|
|
@@ -312,28 +316,130 @@ fig = plotter.plot_stacked(
|
|
|
312
316
|
)
|
|
313
317
|
```
|
|
314
318
|
|
|
315
|
-

|
|
319
|
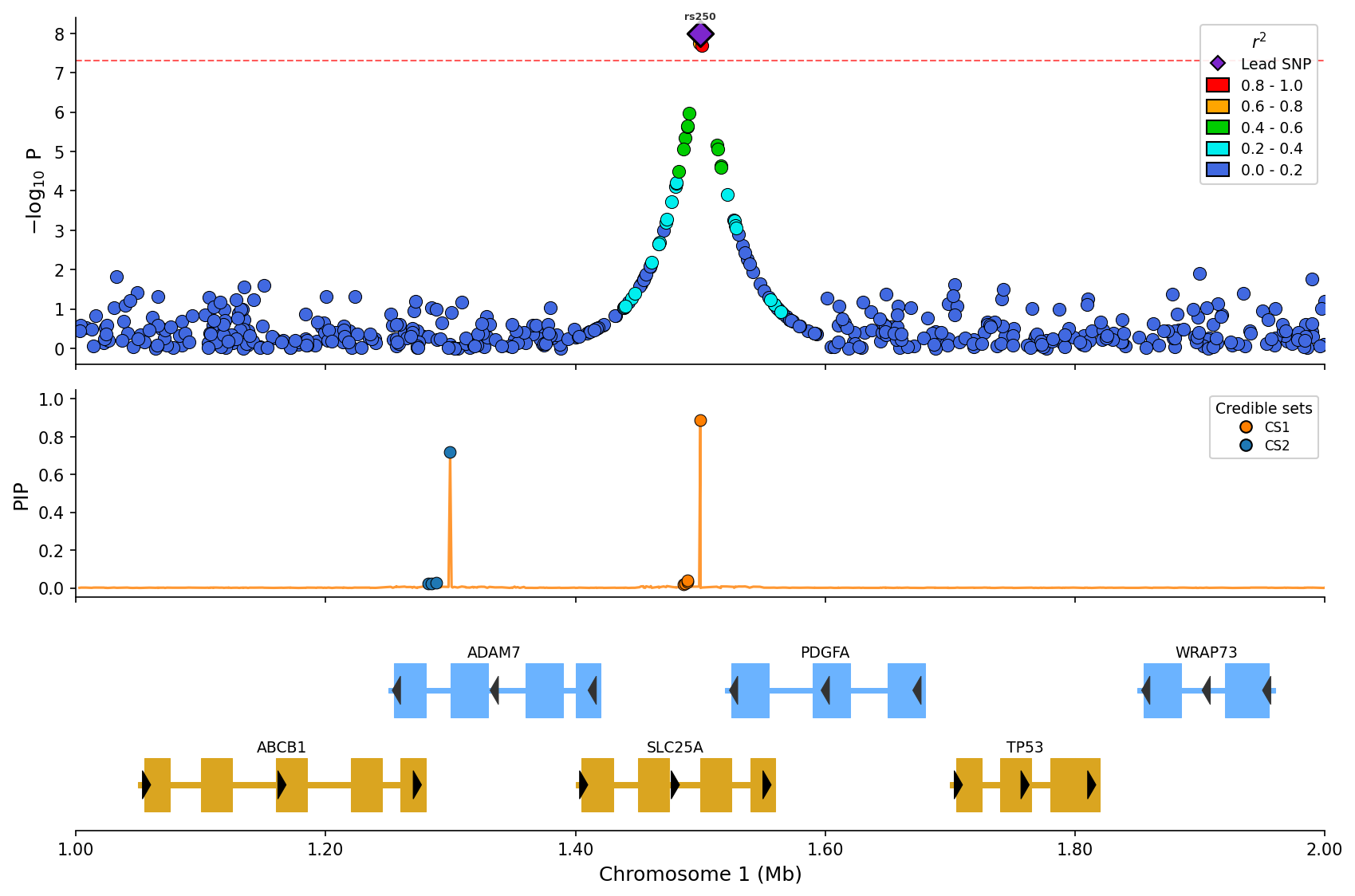
+
|
|
316
320
|
*Fine-mapping visualization with PIP line and credible set coloring (CS1/CS2).*
|
|
317
321
|
|
|
322
|
+
## LD Heatmaps
|
|
323
|
+
|
|
324
|
+
Create triangular LD heatmaps showing pairwise linkage disequilibrium patterns:
|
|
325
|
+
|
|
326
|
+
```python
|
|
327
|
+
from pylocuszoom import LDHeatmapPlotter
|
|
328
|
+
|
|
329
|
+
# ld_matrix is a square DataFrame with SNP IDs as index/columns
|
|
330
|
+
# snp_ids is a list of SNP IDs in the matrix
|
|
331
|
+
|
|
332
|
+
ld_plotter = LDHeatmapPlotter()
|
|
333
|
+
fig = ld_plotter.plot(
|
|
334
|
+
ld_matrix,
|
|
335
|
+
snp_ids,
|
|
336
|
+
highlight_snp_id="rs12345", # Highlight lead SNP
|
|
337
|
+
metric="r2", # or "dprime"
|
|
338
|
+
)
|
|
339
|
+
fig.savefig("ld_heatmap.png", dpi=150)
|
|
340
|
+
```
|
|
341
|
+
|
|
342
|
+
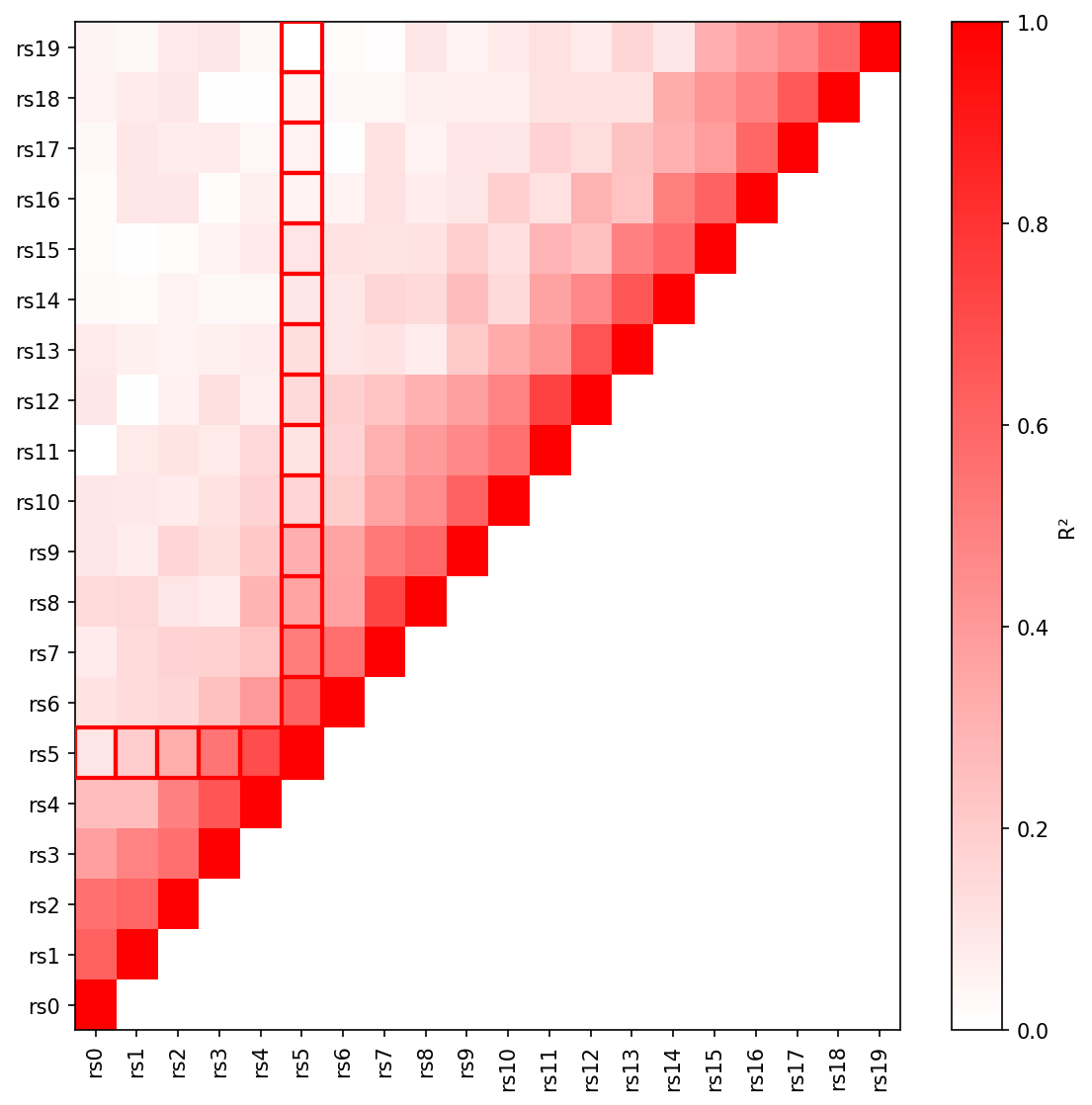
|
|
343
|
+
*Triangular LD heatmap with R² values and lead SNP highlighted.*
|
|
344
|
+
|
|
345
|
+
### Integrated LD Heatmap with Regional Plot
|
|
346
|
+
|
|
347
|
+
Add an LD heatmap panel below a regional association plot:
|
|
348
|
+
|
|
349
|
+
```python
|
|
350
|
+
from pylocuszoom import LocusZoomPlotter
|
|
351
|
+
|
|
352
|
+
plotter = LocusZoomPlotter(species="canine")
|
|
353
|
+
|
|
354
|
+
fig = plotter.plot(
|
|
355
|
+
gwas_df,
|
|
356
|
+
chrom=1,
|
|
357
|
+
start=1000000,
|
|
358
|
+
end=2000000,
|
|
359
|
+
lead_pos=1500000,
|
|
360
|
+
ld_heatmap_df=ld_matrix, # Pairwise LD matrix
|
|
361
|
+
ld_heatmap_snp_ids=snp_ids, # SNP IDs in matrix
|
|
362
|
+
ld_heatmap_height=0.25, # Panel height ratio
|
|
363
|
+
)
|
|
364
|
+
```
|
|
365
|
+
|
|
366
|
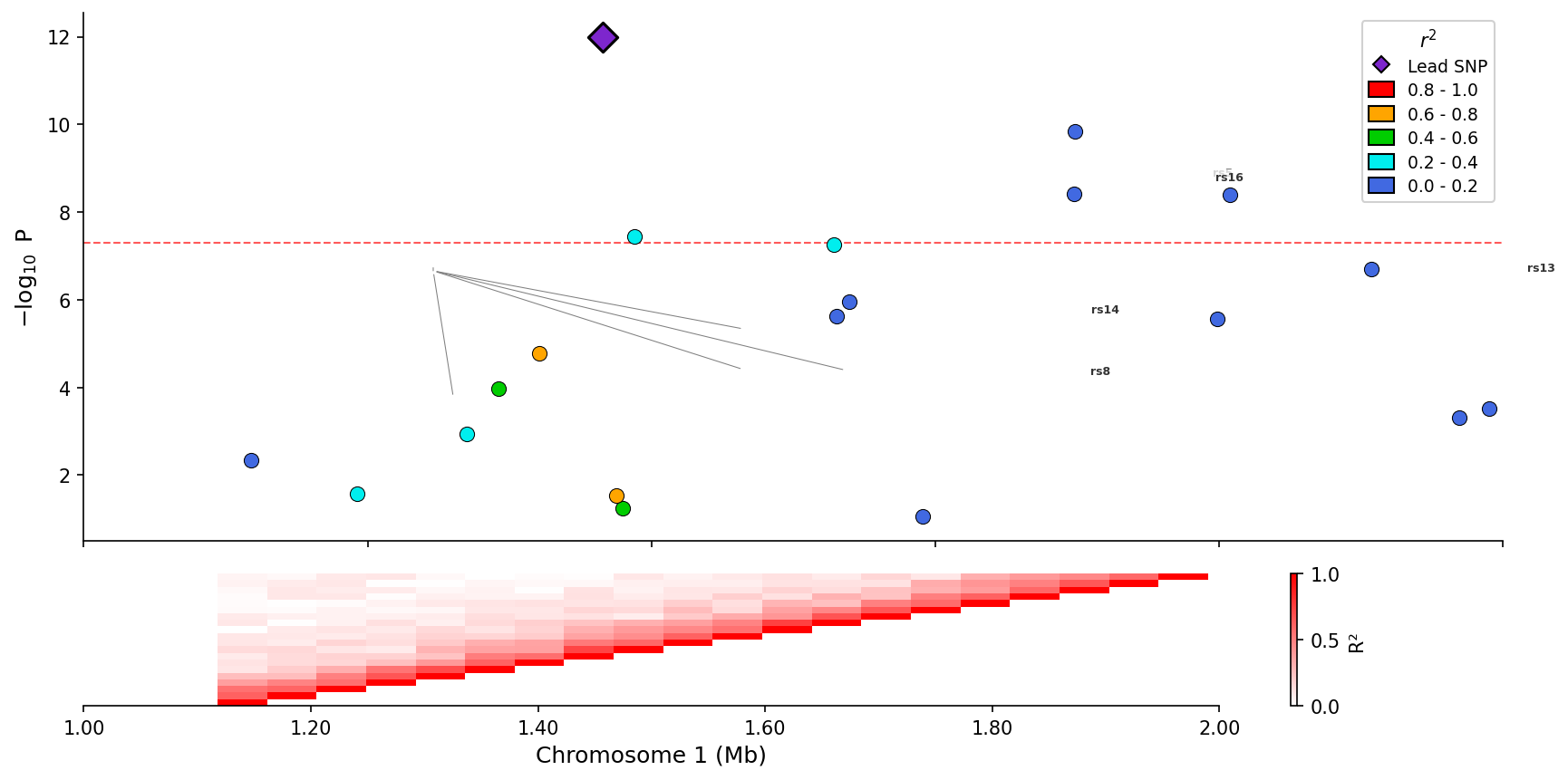
+
|
|
367
|
+
*Regional association plot with integrated LD heatmap panel below.*
|
|
368
|
+
|
|
369
|
+
## Colocalization Plots
|
|
370
|
+
|
|
371
|
+
Visualize GWAS-eQTL colocalization by comparing association signals in a scatter plot with LD coloring:
|
|
372
|
+
|
|
373
|
+
```python
|
|
374
|
+
from pylocuszoom import ColocPlotter
|
|
375
|
+
|
|
376
|
+
# GWAS and eQTL data with matching positions
|
|
377
|
+
gwas_df = pd.DataFrame({
|
|
378
|
+
"pos": positions,
|
|
379
|
+
"p": gwas_pvalues,
|
|
380
|
+
"ld_r2": ld_values, # Optional: LD with lead SNP
|
|
381
|
+
})
|
|
382
|
+
|
|
383
|
+
eqtl_df = pd.DataFrame({
|
|
384
|
+
"pos": positions,
|
|
385
|
+
"p": eqtl_pvalues,
|
|
386
|
+
})
|
|
387
|
+
|
|
388
|
+
plotter = ColocPlotter()
|
|
389
|
+
fig = plotter.plot_coloc(
|
|
390
|
+
gwas_df=gwas_df,
|
|
391
|
+
eqtl_df=eqtl_df,
|
|
392
|
+
pos_col="pos",
|
|
393
|
+
gwas_p_col="p",
|
|
394
|
+
eqtl_p_col="p",
|
|
395
|
+
ld_col="ld_r2",
|
|
396
|
+
gwas_threshold=5e-8,
|
|
397
|
+
eqtl_threshold=1e-5,
|
|
398
|
+
)
|
|
399
|
+
fig.savefig("colocalization.png", dpi=150)
|
|
400
|
+
```
|
|
401
|
+
|
|
402
|
+
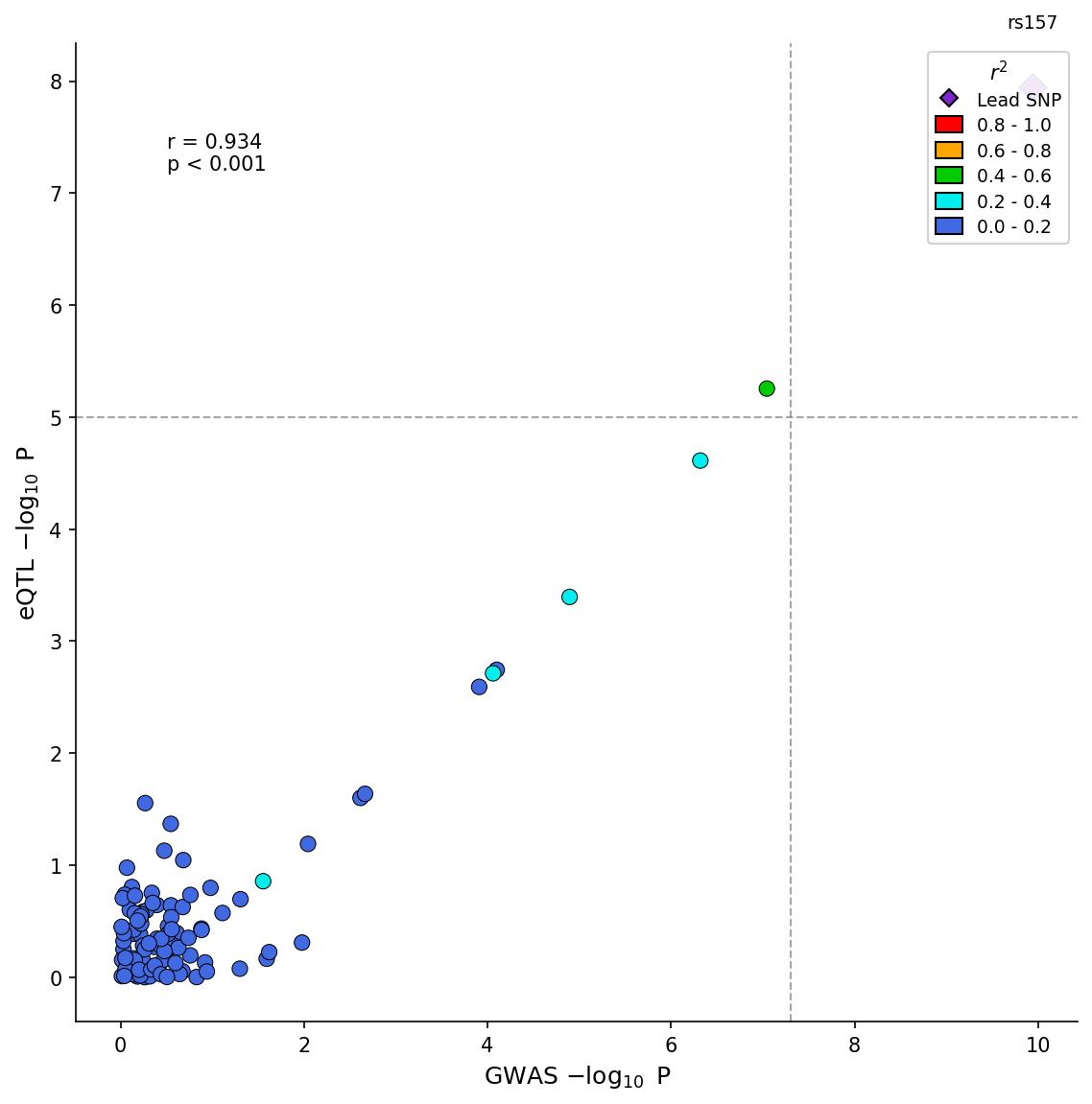
|
|
403
|
+
*GWAS-eQTL colocalization scatter plot with LD coloring and correlation statistics.*
|
|
404
|
+
|
|
405
|
+
**Advanced options** include effect direction coloring and H4 posterior probability display:
|
|
406
|
+
|
|
407
|
+
```python
|
|
408
|
+
fig = plotter.plot_coloc(
|
|
409
|
+
gwas_df=gwas_df,
|
|
410
|
+
eqtl_df=eqtl_df,
|
|
411
|
+
pos_col="pos",
|
|
412
|
+
gwas_p_col="p",
|
|
413
|
+
eqtl_p_col="p",
|
|
414
|
+
gwas_effect_col="beta",
|
|
415
|
+
eqtl_effect_col="slope",
|
|
416
|
+
color_by_effect=True, # Green=congruent, Red=incongruent
|
|
417
|
+
h4_posterior=0.85, # Display coloc H4 probability
|
|
418
|
+
)
|
|
419
|
+
```
|
|
420
|
+
|
|
318
421
|
## PheWAS Plots
|
|
319
422
|
|
|
320
423
|
Visualize associations of a single variant across multiple phenotypes:
|
|
321
424
|
|
|
322
425
|
```python
|
|
426
|
+
from pylocuszoom import StatsPlotter
|
|
427
|
+
|
|
323
428
|
phewas_df = pd.DataFrame({
|
|
324
429
|
"phenotype": ["Height", "BMI", "T2D", "CAD", "HDL"],
|
|
325
430
|
"p_value": [1e-15, 0.05, 1e-8, 1e-3, 1e-10],
|
|
326
431
|
"category": ["Anthropometric", "Anthropometric", "Metabolic", "Cardiovascular", "Lipids"],
|
|
327
432
|
})
|
|
328
433
|
|
|
329
|
-
|
|
434
|
+
stats_plotter = StatsPlotter()
|
|
435
|
+
fig = stats_plotter.plot_phewas(
|
|
330
436
|
phewas_df,
|
|
331
437
|
variant_id="rs12345",
|
|
332
438
|
category_col="category",
|
|
333
439
|
)
|
|
334
440
|
```
|
|
335
441
|
|
|
336
|
-

|
|
442
|
+
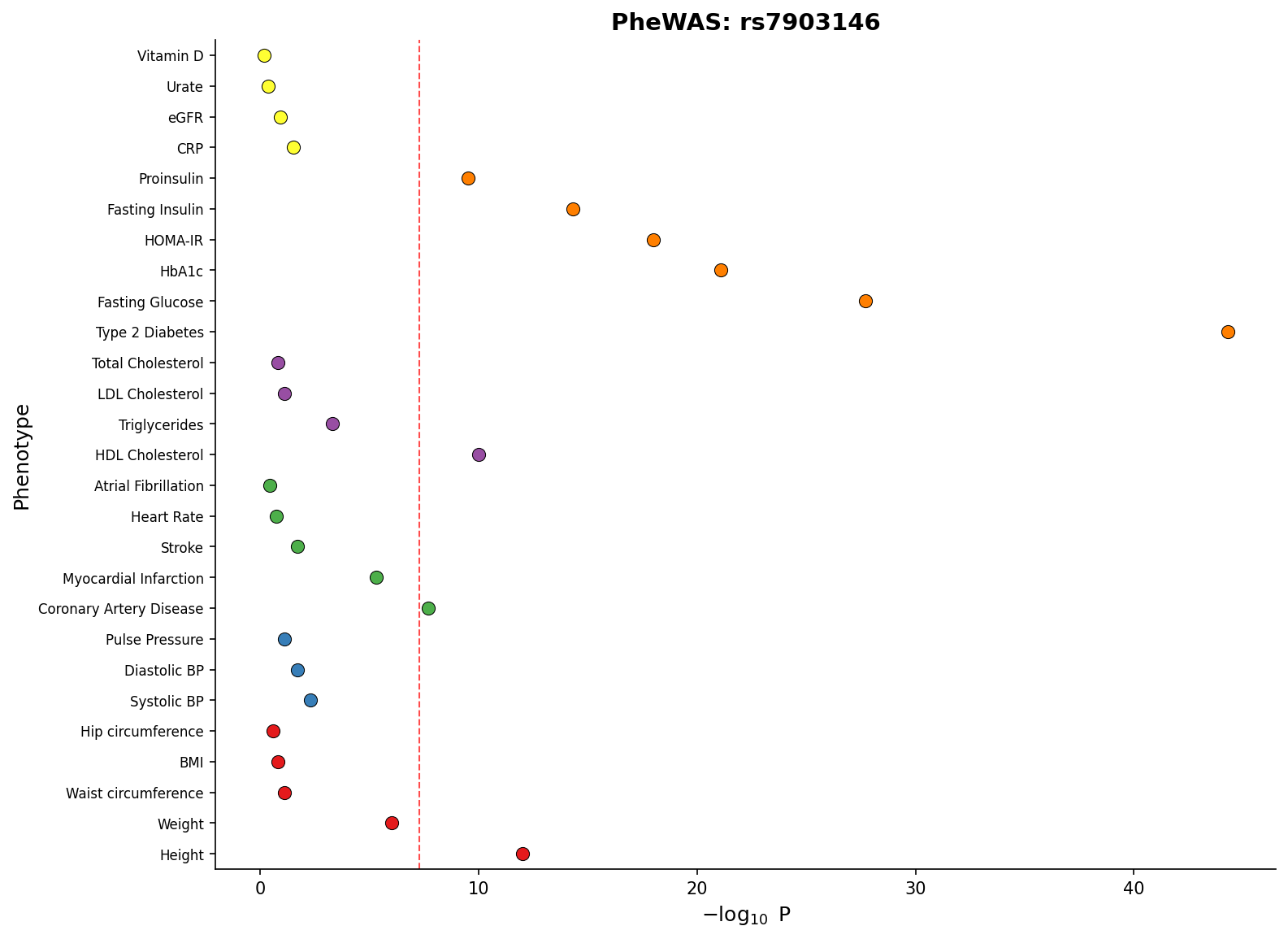
|
|
337
443
|
*PheWAS plot showing associations across phenotype categories with significance threshold.*
|
|
338
444
|
|
|
339
445
|
## Forest Plots
|
|
@@ -341,6 +447,8 @@ fig = plotter.plot_phewas(
|
|
|
341
447
|
Create forest plots for meta-analysis visualization:
|
|
342
448
|
|
|
343
449
|
```python
|
|
450
|
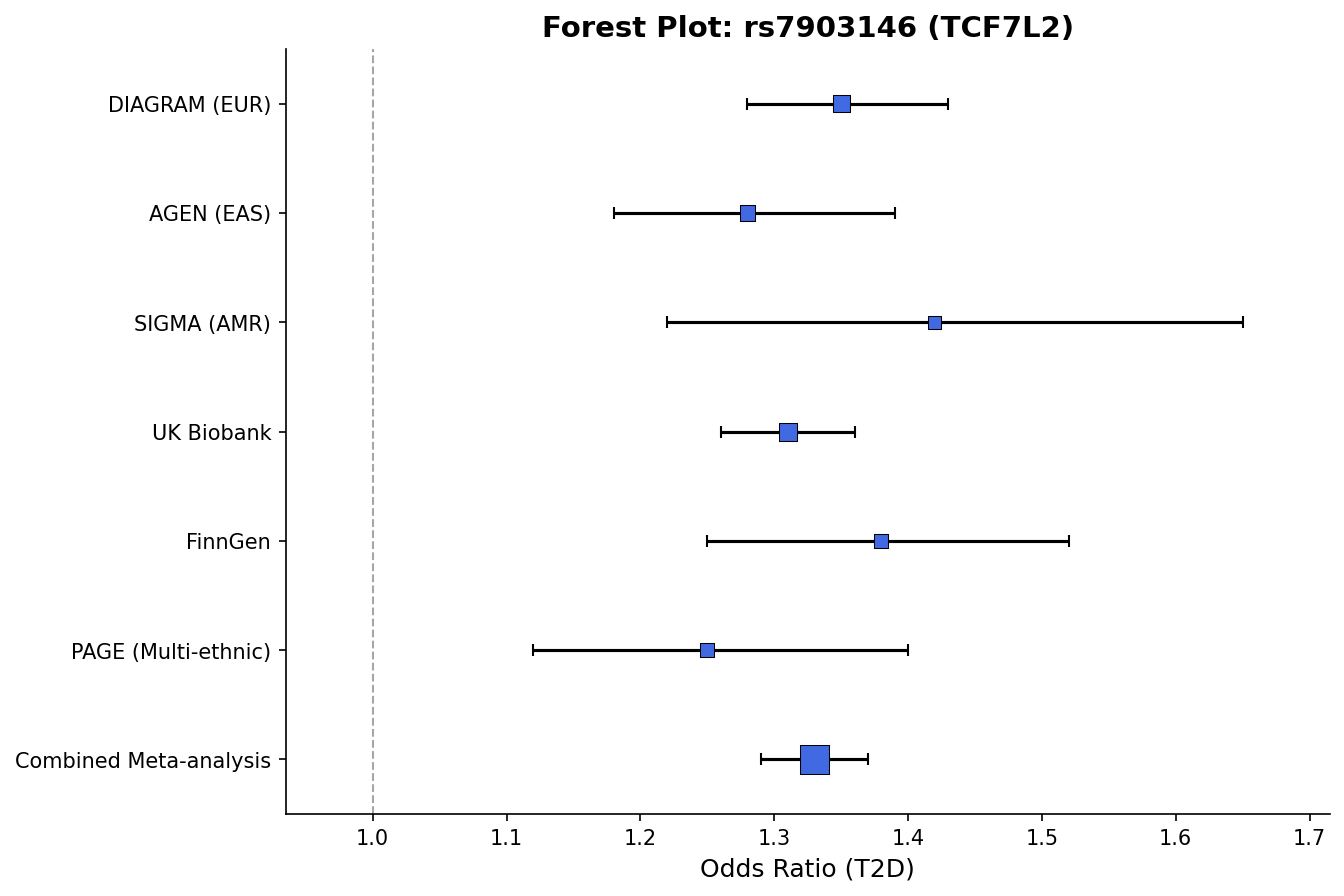
+
from pylocuszoom import StatsPlotter
|
|
451
|
+
|
|
344
452
|
forest_df = pd.DataFrame({
|
|
345
453
|
"study": ["Study A", "Study B", "Study C", "Meta-analysis"],
|
|
346
454
|
"effect": [0.45, 0.52, 0.38, 0.46],
|
|
@@ -349,24 +457,68 @@ forest_df = pd.DataFrame({
|
|
|
349
457
|
"weight": [25, 35, 20, 100],
|
|
350
458
|
})
|
|
351
459
|
|
|
352
|
-
|
|
460
|
+
stats_plotter = StatsPlotter()
|
|
461
|
+
fig = stats_plotter.plot_forest(
|
|
353
462
|
forest_df,
|
|
354
463
|
variant_id="rs12345",
|
|
355
464
|
weight_col="weight",
|
|
356
465
|
)
|
|
357
466
|
```
|
|
358
467
|
|
|
359
|
-

|
|
468
|
+
|
|
360
469
|
*Forest plot with effect sizes, confidence intervals, and weight-proportional markers.*
|
|
361
470
|
|
|
471
|
+
## Miami Plots
|
|
472
|
+
|
|
473
|
+
Compare two GWAS datasets with mirrored Manhattan plots (top panel ascending, bottom panel inverted):
|
|
474
|
+
|
|
475
|
+
```python
|
|
476
|
+
from pylocuszoom import MiamiPlotter
|
|
477
|
+
|
|
478
|
+
plotter = MiamiPlotter(species="human")
|
|
479
|
+
|
|
480
|
+
fig = plotter.plot_miami(
|
|
481
|
+
discovery_df,
|
|
482
|
+
replication_df,
|
|
483
|
+
chrom_col="chrom",
|
|
484
|
+
pos_col="pos",
|
|
485
|
+
p_col="p",
|
|
486
|
+
top_label="Discovery",
|
|
487
|
+
bottom_label="Replication",
|
|
488
|
+
top_threshold=5e-8,
|
|
489
|
+
bottom_threshold=1e-6,
|
|
490
|
+
highlight_regions=[("6", 30_000_000, 35_000_000)], # Highlight MHC region
|
|
491
|
+
)
|
|
492
|
+
fig.savefig("miami.png", dpi=150)
|
|
493
|
+
```
|
|
494
|
+
|
|
495
|
+
**Interactive backends** (Plotly/Bokeh) provide hover tooltips showing SNP details:
|
|
496
|
+
|
|
497
|
+
```python
|
|
498
|
+
# Plotly - interactive HTML with hover tooltips
|
|
499
|
+
plotter = MiamiPlotter(species="human", backend="plotly")
|
|
500
|
+
fig = plotter.plot_miami(discovery_df, replication_df, ...)
|
|
501
|
+
fig.write_html("miami_interactive.html")
|
|
502
|
+
|
|
503
|
+
# Bokeh - dashboard-ready interactive plots
|
|
504
|
+
from bokeh.io import output_file, save
|
|
505
|
+
plotter = MiamiPlotter(species="human", backend="bokeh")
|
|
506
|
+
fig = plotter.plot_miami(discovery_df, replication_df, ...)
|
|
507
|
+
output_file("miami_bokeh.html")
|
|
508
|
+
save(fig)
|
|
509
|
+
```
|
|
510
|
+
|
|
511
|
+
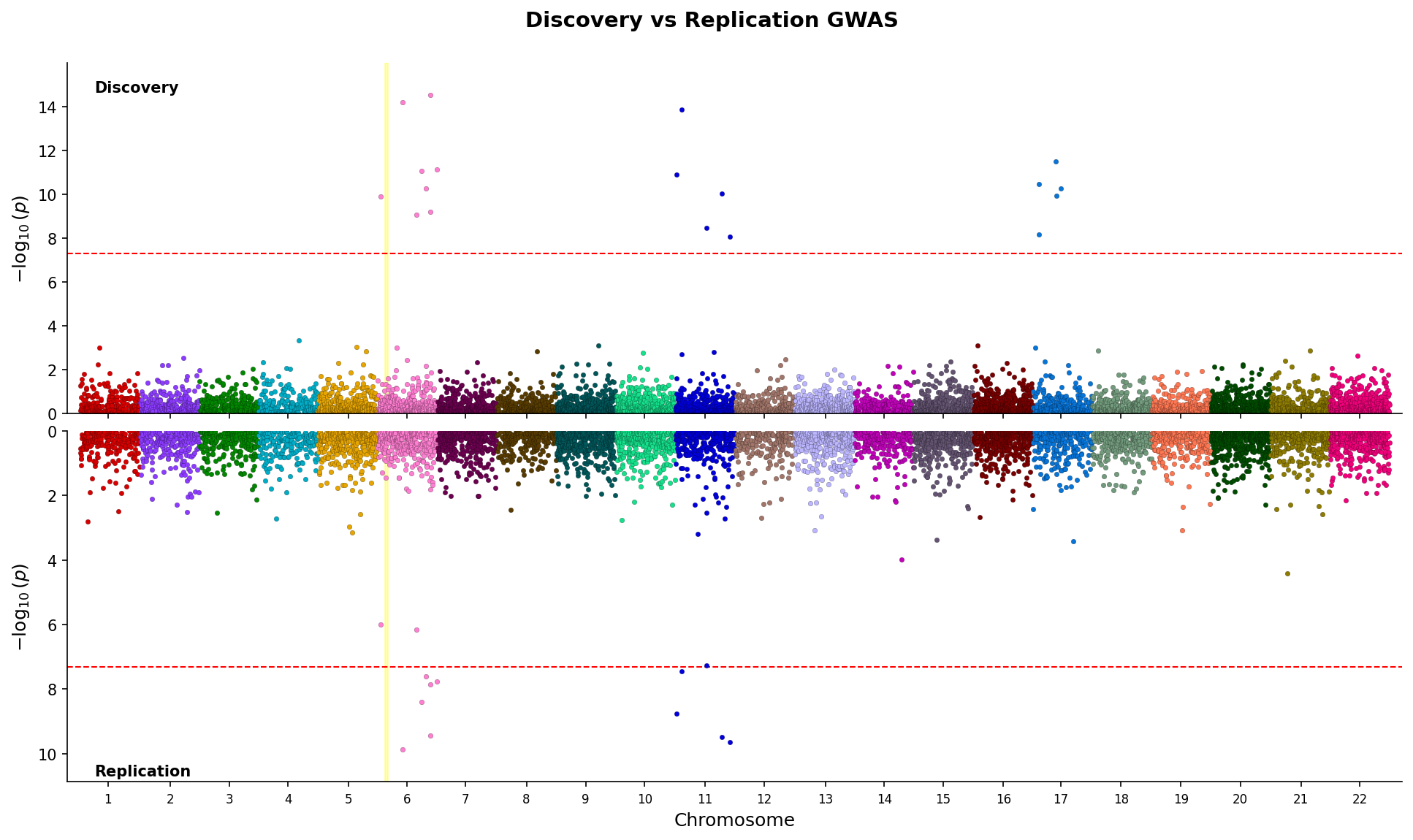
|
|
512
|
+
*Miami plot comparing discovery and replication GWAS with mirrored y-axes and region highlighting.*
|
|
513
|
+
|
|
362
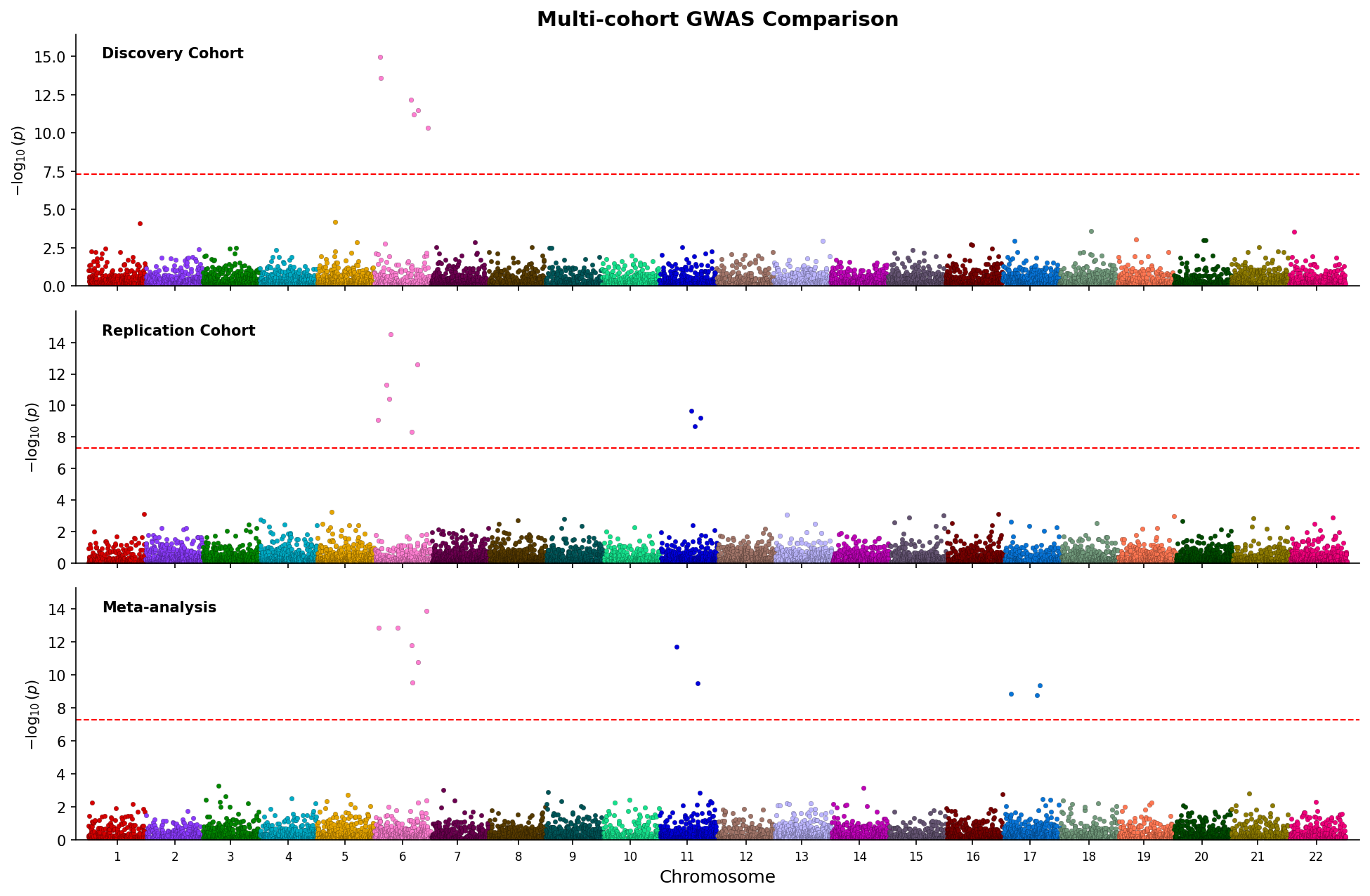
514
|
## Manhattan Plots
|
|
363
515
|
|
|
364
516
|
Create genome-wide Manhattan plots showing associations across all chromosomes:
|
|
365
517
|
|
|
366
518
|
```python
|
|
367
|
-
from pylocuszoom import
|
|
519
|
+
from pylocuszoom import ManhattanPlotter
|
|
368
520
|
|
|
369
|
-
plotter =
|
|
521
|
+
plotter = ManhattanPlotter(species="human")
|
|
370
522
|
|
|
371
523
|
fig = plotter.plot_manhattan(
|
|
372
524
|
gwas_df,
|
|
@@ -379,7 +531,7 @@ fig = plotter.plot_manhattan(
|
|
|
379
531
|
fig.savefig("manhattan.png", dpi=150)
|
|
380
532
|
```
|
|
381
533
|
|
|
382
|
-

|
|
534
|
+
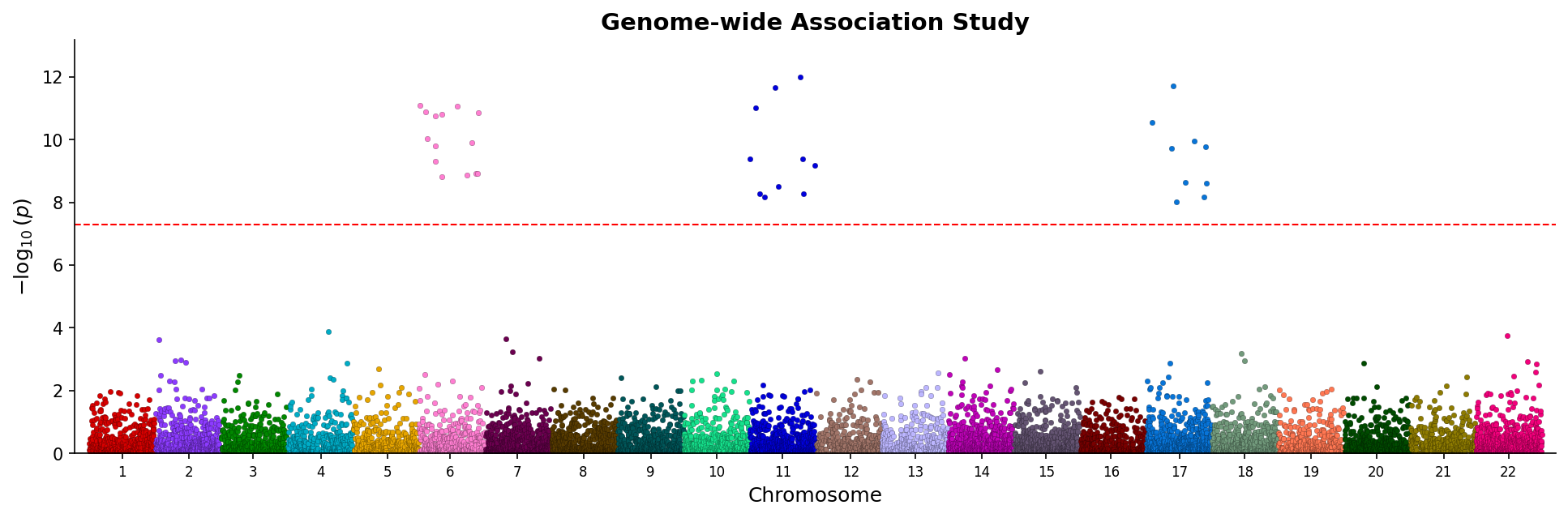
|
|
383
535
|
*Manhattan plot showing genome-wide associations with chromosome coloring and significance threshold.*
|
|
384
536
|
|
|
385
537
|
Categorical Manhattan plots (PheWAS-style) are also supported:
|
|
@@ -397,9 +549,9 @@ fig = plotter.plot_manhattan(
|
|
|
397
549
|
Create quantile-quantile plots to assess p-value distribution:
|
|
398
550
|
|
|
399
551
|
```python
|
|
400
|
-
from pylocuszoom import
|
|
552
|
+
from pylocuszoom import ManhattanPlotter
|
|
401
553
|
|
|
402
|
-
plotter =
|
|
554
|
+
plotter = ManhattanPlotter()
|
|
403
555
|
|
|
404
556
|
fig = plotter.plot_qq(
|
|
405
557
|
gwas_df,
|
|
@@ -411,7 +563,7 @@ fig = plotter.plot_qq(
|
|
|
411
563
|
fig.savefig("qq_plot.png", dpi=150)
|
|
412
564
|
```
|
|
413
565
|
|
|
414
|
-

|
|
566
|
+
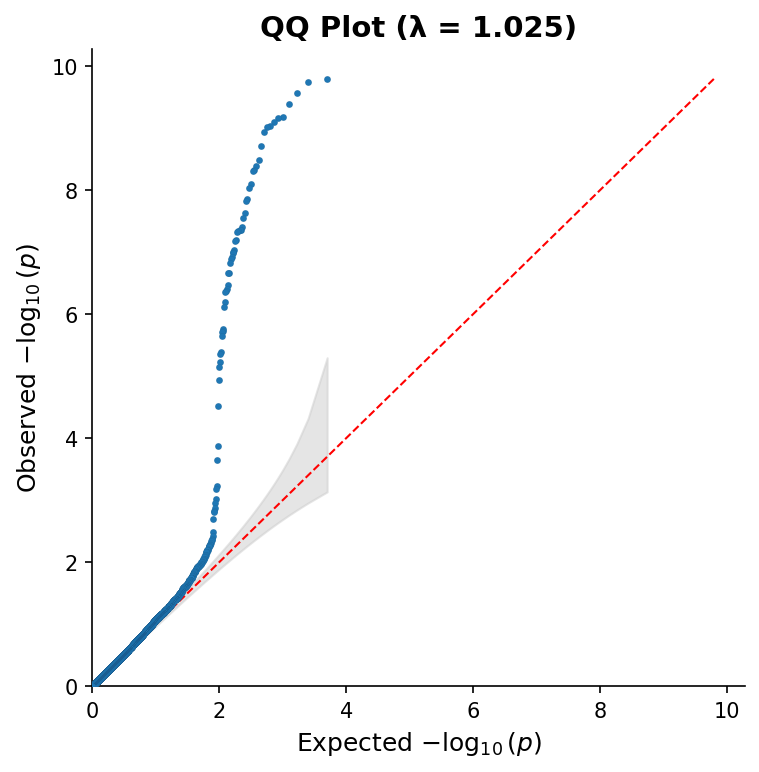
|
|
415
567
|
*QQ plot with 95% confidence band and genomic inflation factor (λ).*
|
|
416
568
|
|
|
417
569
|
## Stacked Manhattan Plots
|
|
@@ -419,9 +571,9 @@ fig.savefig("qq_plot.png", dpi=150)
|
|
|
419
571
|
Compare multiple GWAS results in vertically stacked Manhattan plots:
|
|
420
572
|
|
|
421
573
|
```python
|
|
422
|
-
from pylocuszoom import
|
|
574
|
+
from pylocuszoom import ManhattanPlotter
|
|
423
575
|
|
|
424
|
-
plotter =
|
|
576
|
+
plotter = ManhattanPlotter()
|
|
425
577
|
|
|
426
578
|
fig = plotter.plot_manhattan_stacked(
|
|
427
579
|
[gwas_study1, gwas_study2, gwas_study3],
|
|
@@ -436,7 +588,7 @@ fig = plotter.plot_manhattan_stacked(
|
|
|
436
588
|
fig.savefig("manhattan_stacked.png", dpi=150)
|
|
437
589
|
```
|
|
438
590
|
|
|
439
|
-

|
|
591
|
+
|
|
440
592
|
*Stacked Manhattan plots comparing three GWAS studies with shared chromosome axis.*
|
|
441
593
|
|
|
442
594
|
## Manhattan and QQ Side-by-Side
|
|
@@ -444,9 +596,9 @@ fig.savefig("manhattan_stacked.png", dpi=150)
|
|
|
444
596
|
Create combined Manhattan and QQ plots in a single figure:
|
|
445
597
|
|
|
446
598
|
```python
|
|
447
|
-
from pylocuszoom import
|
|
599
|
+
from pylocuszoom import ManhattanPlotter
|
|
448
600
|
|
|
449
|
-
plotter =
|
|
601
|
+
plotter = ManhattanPlotter()
|
|
450
602
|
|
|
451
603
|
fig = plotter.plot_manhattan_qq(
|
|
452
604
|
gwas_df,
|
|
@@ -462,7 +614,7 @@ fig = plotter.plot_manhattan_qq(
|
|
|
462
614
|
fig.savefig("manhattan_qq.png", dpi=150)
|
|
463
615
|
```
|
|
464
616
|
|
|
465
|
-

|
|
617
|
+
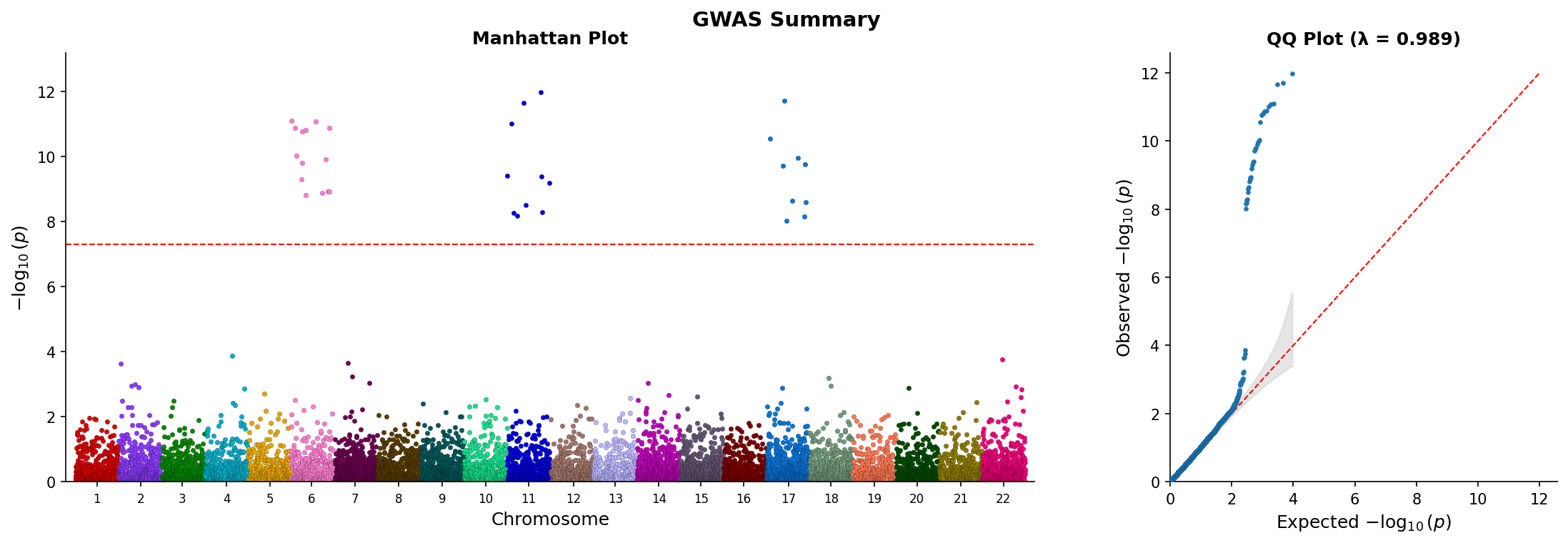
|
|
466
618
|
*Combined Manhattan and QQ plot showing genome-wide associations and p-value distribution.*
|
|
467
619
|
|
|
468
620
|
## PySpark Support
|
|
@@ -1,36 +1,40 @@
|
|
|
1
|
-
pylocuszoom/__init__.py,sha256=
|
|
1
|
+
pylocuszoom/__init__.py,sha256=VwZBIjjY-k6oQVkSgcsAwTkDitHdaHRe2gDUZ5b1YhA,6599
|
|
2
2
|
pylocuszoom/_plotter_utils.py,sha256=ELdSOcKk2KvOo_AxEWHeutmmUS4zZMaDMmQfpQUWaF0,1541
|
|
3
|
-
pylocuszoom/
|
|
4
|
-
pylocuszoom/
|
|
3
|
+
pylocuszoom/coloc.py,sha256=ND56BgUp_yLDTak2RsD9OqZK-X93eDGkL9Q97C2_Fz0,2191
|
|
4
|
+
pylocuszoom/coloc_plotter.py,sha256=9q76_4mYLNU6PjVw_rqx2THdTleI9U6mpzGBA6PnfrY,14036
|
|
5
|
+
pylocuszoom/colors.py,sha256=j1qBHHNTIE9k4bAkIrl7B7bTpTQdWq34-DdJ_y7hZPk,8751
|
|
6
|
+
pylocuszoom/config.py,sha256=Ow8YiW5Yl801K71jOnrPivHvAbA58uH1BvCAbV9hojM,16008
|
|
5
7
|
pylocuszoom/ensembl.py,sha256=w2msgBoIrY79iHI3hURSbevvdFHxHyWF9Z78hXtAaBc,14296
|
|
6
8
|
pylocuszoom/eqtl.py,sha256=9hGcFARABQRCMN3rco0pVlFJdmlh4SLBBKSgOvdIH_U,5924
|
|
7
9
|
pylocuszoom/exceptions.py,sha256=nd-rWMUodW62WVV4TfcYVPQcb66xV6v9FA-_4xHb5VY,926
|
|
8
|
-
pylocuszoom/finemapping.py,sha256=
|
|
10
|
+
pylocuszoom/finemapping.py,sha256=S3ulQj3fkaDM3n4I8EBymbWym_kTD5NEqfIEj93Mdjk,9630
|
|
9
11
|
pylocuszoom/forest.py,sha256=K-wBinxBOqIzsNMtZJ587e_oMhUXIXEqmEzVTUbmHSY,1161
|
|
10
12
|
pylocuszoom/gene_track.py,sha256=Sh0JCSdLNAAH0NQEiDVMvyXjm63PiCMq3gLvewcagvo,17277
|
|
11
|
-
pylocuszoom/labels.py,sha256=
|
|
12
|
-
pylocuszoom/ld.py,sha256=
|
|
13
|
+
pylocuszoom/labels.py,sha256=RPgEcCGPA2VQ1rXIW3WYy0AQ0EE0VBzbh_4EkhsWsNc,4343
|
|
14
|
+
pylocuszoom/ld.py,sha256=CphRau26XL9MoHiU3qRXlSz1Eg39TJt7MNIPZSZDr8M,13999
|
|
15
|
+
pylocuszoom/ld_heatmap_plotter.py,sha256=siHKL-tq7qQBTgyfH09uL0YJPmOGxG3udq8p1We5p0I,8373
|
|
13
16
|
pylocuszoom/loaders.py,sha256=KpWPBO0BCb2yrGTtgdiOqOuhx2YLmjK_ywmpr3onnx8,25156
|
|
14
17
|
pylocuszoom/logging.py,sha256=nZHEkbnjp8zoyWj_S-Hy9UQvUYLoMoxyiOWRozBT2dg,4987
|
|
15
18
|
pylocuszoom/manhattan.py,sha256=sNhPnsfsIqe1ls74D-kKMFyF_ZmaYB9Ul8qf4UMWnF0,8022
|
|
16
19
|
pylocuszoom/manhattan_plotter.py,sha256=1QQxaXEh5YG4x6ZIxpdhdfQPI2KuO_525qYKI7c32n4,27584
|
|
20
|
+
pylocuszoom/miami_plotter.py,sha256=J7GcgeIKyJvJTpQQHx9YNwH5NzpYSej2HPFSGu5YeLY,18099
|
|
17
21
|
pylocuszoom/phewas.py,sha256=6g2LmwA5kmxYlHgPxJvuXIMerEqfqgsrth110Y3CgVU,968
|
|
18
|
-
pylocuszoom/plotter.py,sha256=
|
|
22
|
+
pylocuszoom/plotter.py,sha256=I6fktn1mwE-ZM6bnKUqXrdN_eMQ6oHGaJBq9DwoT4xI,60488
|
|
19
23
|
pylocuszoom/py.typed,sha256=47DEQpj8HBSa-_TImW-5JCeuQeRkm5NMpJWZG3hSuFU,0
|
|
20
24
|
pylocuszoom/qq.py,sha256=GPIFHXYCLvhP4IUgjcU3QELLREH8r1AEYXMord8gtEo,3650
|
|
21
|
-
pylocuszoom/recombination.py,sha256=
|
|
25
|
+
pylocuszoom/recombination.py,sha256=M-wDBdGbC5qGDHPFoGzBTPmTdiRD7bpRskyxBAKxTUY,15878
|
|
22
26
|
pylocuszoom/schemas.py,sha256=XxeivyRm5LGDwJw4GToxzOSdyx1yXvFYk3xgeFJ6VW0,11858
|
|
23
27
|
pylocuszoom/stats_plotter.py,sha256=67bgU-TXGnmVTxfTRWT3-PFemVVy6lTu4-ZlxUnwHS4,11171
|
|
24
28
|
pylocuszoom/utils.py,sha256=Z2P__Eau3ilF2ftuAZBm11EZ1NqCFQzfr4br9jCiJmg,6887
|
|
25
29
|
pylocuszoom/validation.py,sha256=3D9axjUvNXWW3Mk7dwRG38-di2P0zDpVVGF5WNSfZbk,7403
|
|
26
30
|
pylocuszoom/backends/__init__.py,sha256=xefVj3jVxmYwVLLY5AZtFqTPMehQxZ2qGd-Pk7_V_Bk,4267
|
|
27
|
-
pylocuszoom/backends/base.py,sha256=
|
|
28
|
-
pylocuszoom/backends/bokeh_backend.py,sha256=
|
|
31
|
+
pylocuszoom/backends/base.py,sha256=Wgzr6ncKnqZpjEYrc6aPmIKDWdZLm18mMO7q7XRU6SA,25640
|
|
32
|
+
pylocuszoom/backends/bokeh_backend.py,sha256=ft0W5ScxPoA6T6GB7k9PFfTMgOCyFom_plqVItdaUA0,34198
|
|
29
33
|
pylocuszoom/backends/hover.py,sha256=Hjm_jcxJL8dDxO_Ye7jeWAUcHKlbH6oO8ZfGJ2MzIFM,6564
|
|
30
|
-
pylocuszoom/backends/matplotlib_backend.py,sha256=
|
|
31
|
-
pylocuszoom/backends/plotly_backend.py,sha256=
|
|
34
|
+
pylocuszoom/backends/matplotlib_backend.py,sha256=SKTL_6QCc2D_UVITH543O-zPD_I8AwDLCZE0Re0KfE0,27269
|
|
35
|
+
pylocuszoom/backends/plotly_backend.py,sha256=cow67G8dIcrxSI3XOPFWJ9hSLqe1-MB4NFSeO8GDRG4,42228
|
|
32
36
|
pylocuszoom/reference_data/__init__.py,sha256=qqHqAUt1jebGlCN3CjqW3Z-_coHVNo5K3a3bb9o83hA,109
|
|
33
|
-
pylocuszoom-1.1.
|
|
34
|
-
pylocuszoom-1.1.
|
|
35
|
-
pylocuszoom-1.1.
|
|
36
|
-
pylocuszoom-1.1.
|
|
37
|
+
pylocuszoom-1.3.1.dist-info/METADATA,sha256=nHe6YBAHx718EndDz71xIukqf_WwJ7Q4yERBkgz9xmo,27020
|
|
38
|
+
pylocuszoom-1.3.1.dist-info/WHEEL,sha256=WLgqFyCfm_KASv4WHyYy0P3pM_m7J5L9k2skdKLirC8,87
|
|
39
|
+
pylocuszoom-1.3.1.dist-info/licenses/LICENSE.md,sha256=bqhD2fIhoqfLdX1lyGNoufIHWE7Q8mU-SyFadwKn4cc,34902
|
|
40
|
+
pylocuszoom-1.3.1.dist-info/RECORD,,
|