nci-cidc-schemas 0.28.1__py2.py3-none-any.whl → 0.28.3__py2.py3-none-any.whl
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- cidc_schemas/__init__.py +1 -1
- cidc_schemas/ngs_pipeline_api/__init__.py +29 -0
- cidc_schemas/ngs_pipeline_api/atacseq/atacseq.md +55 -0
- cidc_schemas/ngs_pipeline_api/atacseq/atacseq_output_API.json +39 -0
- cidc_schemas/ngs_pipeline_api/atacseq/imgs/atacseq.png +0 -0
- cidc_schemas/ngs_pipeline_api/output_API.schema.json +45 -0
- cidc_schemas/ngs_pipeline_api/rna/imgs/RIMA.png +0 -0
- cidc_schemas/ngs_pipeline_api/rna/rna.md +54 -0
- cidc_schemas/ngs_pipeline_api/rna/rna_config.schema.json +39 -0
- cidc_schemas/ngs_pipeline_api/rna/rna_output_API.json +195 -0
- cidc_schemas/ngs_pipeline_api/tcr/imgs/TCRseq.png +0 -0
- cidc_schemas/ngs_pipeline_api/tcr/tcr.md +101 -0
- cidc_schemas/ngs_pipeline_api/wes/imgs/wes.png +0 -0
- cidc_schemas/ngs_pipeline_api/wes/wes.md +46 -0
- cidc_schemas/ngs_pipeline_api/wes/wes_config.schema.json +82 -0
- cidc_schemas/ngs_pipeline_api/wes/wes_output_API.json +503 -0
- cidc_schemas/ngs_pipeline_api/wes/wes_output_API.py +548 -0
- cidc_schemas/ngs_pipeline_api/wes/wes_tumor_only_output_API.json +213 -0
- cidc_schemas/prism/constants.py +2 -0
- cidc_schemas/schemas/assays/components/available_assays.json +11 -0
- cidc_schemas/schemas/assays/components/nulisa_input.json +46 -0
- cidc_schemas/schemas/assays/nulisa_assay.json +63 -0
- cidc_schemas/schemas/templates/assays/nulisa_template.json +88 -0
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/METADATA +1 -1
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/RECORD +29 -9
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/WHEEL +0 -0
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/entry_points.txt +0 -0
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/licenses/LICENSE +0 -0
- {nci_cidc_schemas-0.28.1.dist-info → nci_cidc_schemas-0.28.3.dist-info}/top_level.txt +0 -0
cidc_schemas/__init__.py
CHANGED
|
@@ -0,0 +1,29 @@
|
|
|
1
|
+
# NOTE: this is copied form nci-cidc-ngs-pipeline-api==0.1.25 which is archived
|
|
2
|
+
|
|
3
|
+
import os
|
|
4
|
+
from json import load
|
|
5
|
+
|
|
6
|
+
|
|
7
|
+
# __author__ = """NCI"""
|
|
8
|
+
# __email__ = "nci-cidc-tools-admin@mail.nih.gov"
|
|
9
|
+
# __version__ = "0.1.25"
|
|
10
|
+
|
|
11
|
+
|
|
12
|
+
_API_ENDING = "_output_API.json"
|
|
13
|
+
_BASE_DIR = os.path.dirname(os.path.abspath(__file__))
|
|
14
|
+
_SCHEMA_PATH = os.path.join(_BASE_DIR, "output_API.schema.json")
|
|
15
|
+
|
|
16
|
+
|
|
17
|
+
try:
|
|
18
|
+
with open(_SCHEMA_PATH, "r", encoding="UTF") as f:
|
|
19
|
+
METASCHEMA = load(f)
|
|
20
|
+
except Exception as e: # pylint: disable=broad-except
|
|
21
|
+
raise Exception(f"Failed loading json {_SCHEMA_PATH}") from e
|
|
22
|
+
|
|
23
|
+
OUTPUT_APIS = {}
|
|
24
|
+
for dname, _, files in os.walk(_BASE_DIR):
|
|
25
|
+
for fname in files:
|
|
26
|
+
if fname.endswith(_API_ENDING):
|
|
27
|
+
analysis = fname[: -len(_API_ENDING)]
|
|
28
|
+
with open(os.path.join(dname, fname), "rb") as f:
|
|
29
|
+
OUTPUT_APIS[analysis] = load(f)
|
|
@@ -0,0 +1,55 @@
|
|
|
1
|
+
## CHIPS ATAC-seq Pipeline Description
|
|
2
|
+
|
|
3
|
+
The CHIPS pipeline is designed to perform robust quality control and reproducible processing of the chromatin profile sequencing data derived from ChIP-seq, DNase-seq, and ATAC-seq. The CHIPS pipeline includes procedures such as read alignment, peak calling, motif finding, and putative target prediction. The inputs to the pipeline are FASTQ/BAM format DNA sequence read files. The analysis process is split into three main components: read alignment, quality control and downstream analysis. The pipeline itself is encoded in the workflow language [Snakemake](https://snakemake.readthedocs.io/) and executed in a conda environment using the Google Cloud Compute Engine.
|
|
4
|
+
|
|
5
|
+
The main components of the CHIPs ATAC-seq pipline are:
|
|
6
|
+
|
|
7
|
+
* Read alignment
|
|
8
|
+
* Quality control:
|
|
9
|
+
* Mapped reads
|
|
10
|
+
* Sample contamination from other species.
|
|
11
|
+
* Evolutionary conservation
|
|
12
|
+
* Fraction of reads in peaks (FRIP) and PRC bottleneck (PBC) score
|
|
13
|
+
* Overlapping with union Dnase Hypersensitivy Sites.
|
|
14
|
+
* Number of high-quality peaks,
|
|
15
|
+
* Peak calling using [`MACS2`](https://github.com/macs3-project/MACS) and generating genome browser view [bigwig](https://genome.ucsc.edu/goldenPath/help/bigWig.html) file.
|
|
16
|
+
* Downstream analysis:
|
|
17
|
+
* Peak annotation
|
|
18
|
+
* putative target prediction using [Regulatory Potential](https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-1934-6).
|
|
19
|
+
* Motif enrichment using [`Homer`](http://homer.ucsd.edu/homer/motif/)
|
|
20
|
+
|
|
21
|
+
|
|
22
|
+
### workflow figure for ATAC-seq pipeline
|
|
23
|
+
|
|
24
|
+
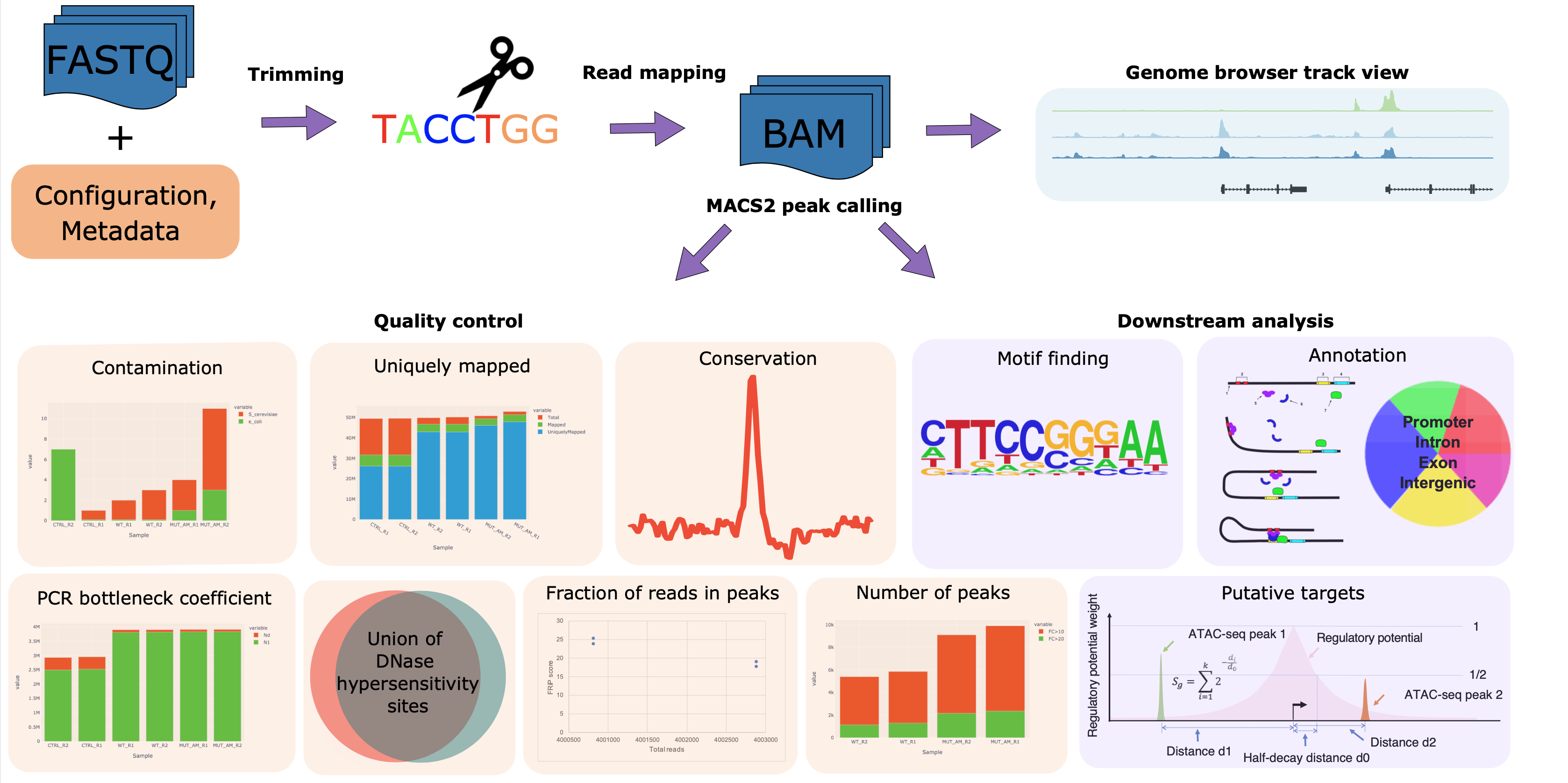
|
|
25
|
+
|
|
26
|
+
## Versions of Tools and Reference Files Used in CHIPs
|
|
27
|
+
|
|
28
|
+
| Software | Version | Source | Notes |
|
|
29
|
+
|------------------|---------|-----------------------|---------------------|
|
|
30
|
+
| snakemake | 5.4.5 | bioconda | Pipeline management |
|
|
31
|
+
| samtools | 1.10 | bioconda | |
|
|
32
|
+
| python | 3.6.12 | conda-forge | |
|
|
33
|
+
| r | 3.5.1 | conda-forge | |
|
|
34
|
+
| numpy | 1.19.5 | conda | |
|
|
35
|
+
| bwa | 0.7.15 | bioconda | Alignment |
|
|
36
|
+
| picard | 2.18.4 | bioconda | Mark duplicates |
|
|
37
|
+
| bedtools | 2.27.1 | bioconda | |
|
|
38
|
+
| seqtk | 1.3 | bioconda | |
|
|
39
|
+
| fastqc | 0.11.9 | bioconda | |
|
|
40
|
+
| ggplot2 | 3.3.0 | conda-forge r | |
|
|
41
|
+
| reshape2 | 1.4.4 | conda-forge r | |
|
|
42
|
+
| git | 2.26.0 | conda-forge | |
|
|
43
|
+
| perl | 5.26.2 | conda-forge | |
|
|
44
|
+
| homer | 4.11 | bioconda | Motif analysis |
|
|
45
|
+
| weblogo | 2.8.2 | bioconda | |
|
|
46
|
+
| seqLogo | 1.50.0 | bioconda bioconductor | |
|
|
47
|
+
| bedgraphtobigwig | 377 | bioconda ucsc | |
|
|
48
|
+
| bedsort | 377 | bioconda ucsc | |
|
|
49
|
+
| seaborn | 0.11.1 | conda-forge | |
|
|
50
|
+
| r.utils | 2.9.2 | conda-forge r | |
|
|
51
|
+
| pybigwig | 0.3.17 | bioconda | |
|
|
52
|
+
| cython | 0.29.2 | conda | |
|
|
53
|
+
| jinja2 | 2.11.2 | conda | |
|
|
54
|
+
| macs2 | 2.2.7 | bioconda | Peak calling |
|
|
55
|
+
| fastp | 0.20.1 | bioconda | Adaptor trimming |
|
|
@@ -0,0 +1,39 @@
|
|
|
1
|
+
{
|
|
2
|
+
"cimac id": [
|
|
3
|
+
{
|
|
4
|
+
"filter_group": "peaks/sorted_peaks",
|
|
5
|
+
"file_path_template": "analysis/peaks/{cimac id}.rep1/{cimac id}.rep1_sorted_peaks.bed",
|
|
6
|
+
"short_description": "Regular peak called by MACS2",
|
|
7
|
+
"long_description": "5th: integer score for display. It's calculated as int(-10*log10pvalue) or int(-10*log10qvalue) depending on whether -p (pvalue) or -q (qvalue) is used as score cutoff 7th: fold-change at peak summit 8th: -log10pvalue at peak summit 9th: -log10qvalue at peak summit 10th: relative summit position to peak start. https://github.com/macs3-project/MACS",
|
|
8
|
+
"file_purpose": "Analysis view"
|
|
9
|
+
},
|
|
10
|
+
{
|
|
11
|
+
"filter_group": "peaks/sorted_summits",
|
|
12
|
+
"file_path_template": "analysis/peaks/{cimac id}.rep1/{cimac id}.rep1_sorted_summits.bed",
|
|
13
|
+
"short_description": "Peak summit called by MACS2",
|
|
14
|
+
"long_description": "MACS2-called location with the highest fragment pileup aka the summit",
|
|
15
|
+
"file_purpose": "Analysis view"
|
|
16
|
+
},
|
|
17
|
+
{
|
|
18
|
+
"filter_group": "peaks/sorted_narrowPeak",
|
|
19
|
+
"file_path_template": "analysis/peaks/{cimac id}.rep1/{cimac id}.rep1_sorted_peaks.narrowPeak",
|
|
20
|
+
"short_description": "narrowPeak called by MACS2",
|
|
21
|
+
"long_description": "MACS2-called peak locations, summits, p-, and q-values in BED6+4 format",
|
|
22
|
+
"file_purpose": "Analysis view"
|
|
23
|
+
},
|
|
24
|
+
{
|
|
25
|
+
"filter_group": "peaks/bigwig",
|
|
26
|
+
"file_path_template": "analysis/peaks/{cimac id}.rep1/{cimac id}.rep1_treat_pileup.bw",
|
|
27
|
+
"short_description": "bigwig file",
|
|
28
|
+
"long_description": "RPKM (reads per kilobase per million) normalized pile up bigwig file for visualization in IGV",
|
|
29
|
+
"file_purpose": "Analysis view"
|
|
30
|
+
},
|
|
31
|
+
{
|
|
32
|
+
"filter_group": "align/sorted_bam",
|
|
33
|
+
"file_path_template": "analysis/align/{cimac id}/{cimac id}.sorted.bam",
|
|
34
|
+
"short_description": "alignment file",
|
|
35
|
+
"long_description": "bwa-mem aligned sorted alignment file",
|
|
36
|
+
"file_purpose": "Source view"
|
|
37
|
+
}
|
|
38
|
+
]
|
|
39
|
+
}
|
|
Binary file
|
|
@@ -0,0 +1,45 @@
|
|
|
1
|
+
{
|
|
2
|
+
"$schema": "http://json-schema.org/draft-07/schema#",
|
|
3
|
+
"$id": "output_API.schema",
|
|
4
|
+
"title": "Pipeline output_API.JSON schema",
|
|
5
|
+
"type": "object",
|
|
6
|
+
"description": "Schema for pipeline's output_API.JSONs",
|
|
7
|
+
"properties": {
|
|
8
|
+
"id": {"type":"null"}
|
|
9
|
+
},
|
|
10
|
+
"additionalProperties": {
|
|
11
|
+
"type": "array",
|
|
12
|
+
"items": {
|
|
13
|
+
"type": "object",
|
|
14
|
+
"properties": {
|
|
15
|
+
"filter_group": {
|
|
16
|
+
"description": "Filter under which the file would appear during faceted search. It is the GCS-URI top-level hierarchy.",
|
|
17
|
+
"type": "string"
|
|
18
|
+
},
|
|
19
|
+
"file_path_template": {
|
|
20
|
+
"description": "Local file path template string, describes where the CLI expects a file to be located during upload.",
|
|
21
|
+
"type": "string"
|
|
22
|
+
},
|
|
23
|
+
"short_description": {
|
|
24
|
+
"description": "Short description, to appear on hover, not more than a sentence long.",
|
|
25
|
+
"type": "string"
|
|
26
|
+
},
|
|
27
|
+
"long_description": {
|
|
28
|
+
"description": "Long description, to appear in file documentation page, 3-5 sentences long.",
|
|
29
|
+
"type": "string"
|
|
30
|
+
},
|
|
31
|
+
"file_purpose": {
|
|
32
|
+
"description": "Assigns a tag to for the file to show up in a particular file-browser view configuration.",
|
|
33
|
+
"type": "string",
|
|
34
|
+
"enum": [
|
|
35
|
+
"Source view",
|
|
36
|
+
"Analysis view",
|
|
37
|
+
"Clinical view",
|
|
38
|
+
"Miscellaneous"
|
|
39
|
+
]
|
|
40
|
+
}
|
|
41
|
+
}
|
|
42
|
+
}
|
|
43
|
+
}
|
|
44
|
+
}
|
|
45
|
+
|
|
Binary file
|
|
@@ -0,0 +1,54 @@
|
|
|
1
|
+
## RIMA (RNA-Seq IMmune Analysis) Pipeline Description
|
|
2
|
+
|
|
3
|
+
Tumor RNA-seq has become an important technique for molecular profiling and
|
|
4
|
+
immune characterization of tumors. RIMA (RNA-seq IMmune Analysis) performs
|
|
5
|
+
integrative computational modeling of tumor microenvironment from bulk tumor
|
|
6
|
+
RNA-seq data, which has the potential to offer essential insights to cancer
|
|
7
|
+
immunology and immune-oncology studies.
|
|
8
|
+
The pre-processing module includes four main procedures:
|
|
9
|
+
- Read mapping
|
|
10
|
+
- Quality control
|
|
11
|
+
- Gene quantification
|
|
12
|
+
- Batch effect removal
|
|
13
|
+
|
|
14
|
+
The downstream analysis includes seven modules:
|
|
15
|
+
- Differential gene expression
|
|
16
|
+
- Immune infiltration estimation
|
|
17
|
+
- Immune repertoire estimation
|
|
18
|
+
- Gene fusion
|
|
19
|
+
- Immunotherapy response prediction
|
|
20
|
+
- HLA prediction
|
|
21
|
+
- Microbiome characterization
|
|
22
|
+
|
|
23
|
+
RIMA uses a conda virtual environment for software compiling and the python
|
|
24
|
+
Snakemake workflow management system for automatic batch processing.
|
|
25
|
+
|
|
26
|
+
### Workflow figure of RIMA pipeline
|
|
27
|
+
|
|
28
|
+
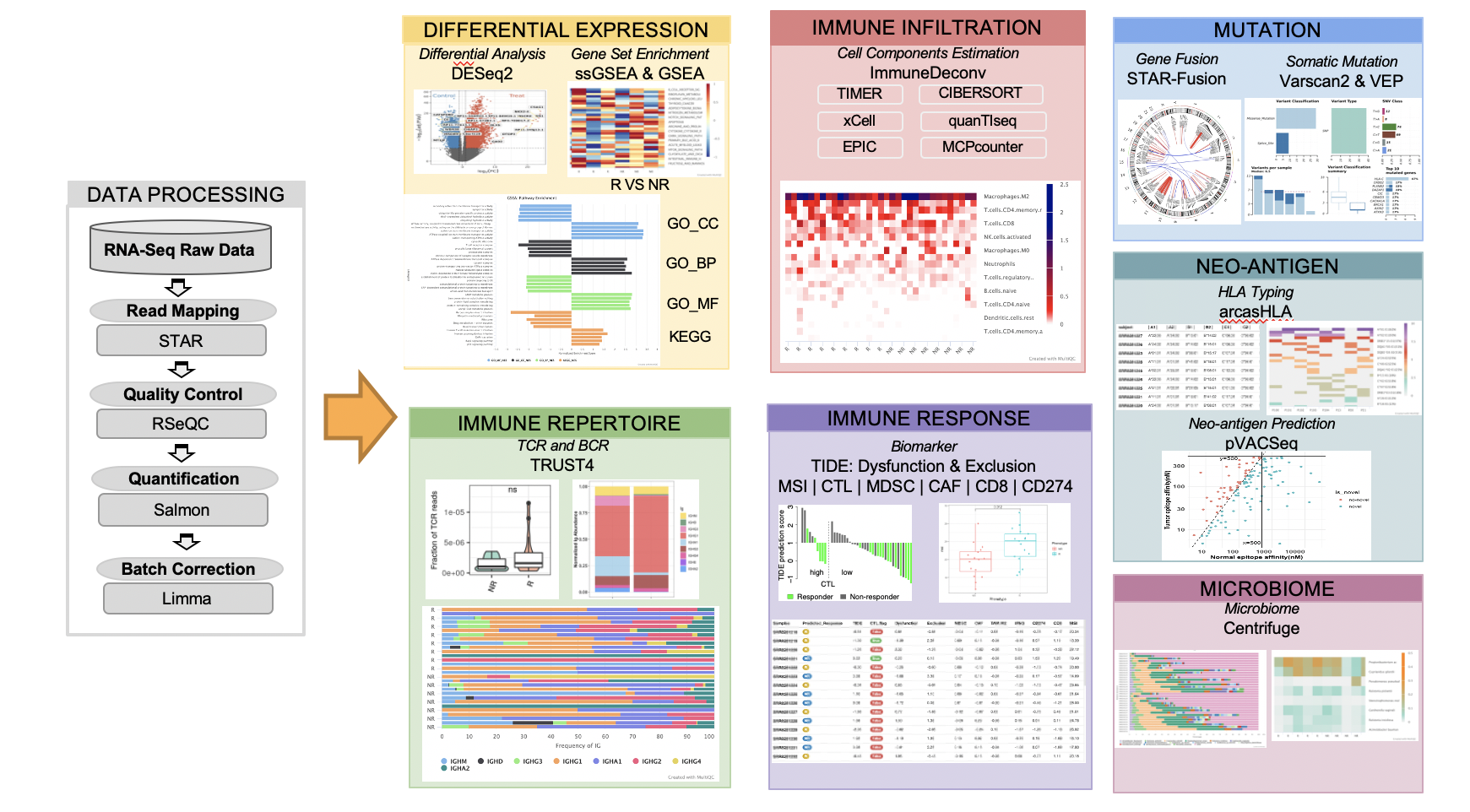
|
|
29
|
+
|
|
30
|
+
### Versions of Tools used in RIMA
|
|
31
|
+
|
|
32
|
+
| Software | Version | Source | Notes |
|
|
33
|
+
|:-----------------|:--------|:----------------------|:----------------------------------|
|
|
34
|
+
| conda | 4.8.3 | bioconda | Environment Management |
|
|
35
|
+
| Snakemake | 5.5.4 | bioconda | Pipeline Management |
|
|
36
|
+
| python | 3.6.8 | conda-forge | Scripting Language |
|
|
37
|
+
| numpy | 1.19.1 | conda-forge | python library |
|
|
38
|
+
| perl | 5.26.2 | conda-forge | Scripting Language |
|
|
39
|
+
| r | 3.5.1 | conda-forge | Scripting Language |
|
|
40
|
+
| star | 2.6.1 | bioconda | Read Alignment |
|
|
41
|
+
| star-fusion | 1.5.0 | bioconda | Fusion Transcripts |
|
|
42
|
+
| samtools | 1.9 | bioconda | Alignment Utility |
|
|
43
|
+
| salmon | 1.3.0 | bioconda | Gene Quantification |
|
|
44
|
+
| picard | 2.20.4 | bioconda | Utility tool for aligned files |
|
|
45
|
+
| bedtools | 2.26.0 | bioconda | Utility tool for aligned files |
|
|
46
|
+
| bcftools | 1.9 | bioconda | BCF Manipulation |
|
|
47
|
+
| rseqc | 3.0.1 | bioconda | Quality Check |
|
|
48
|
+
| limma | 3.42.0 | bioconductor | Batch Removal |
|
|
49
|
+
| deseq2 | 1.26.0 | bioconductor | Differential Expression Analysis |
|
|
50
|
+
| MSISensor2 | 0.1 | git | Microsatellite Instability |
|
|
51
|
+
| TRUST4 | 1.0.0 | git | Immune repertoire analysis |
|
|
52
|
+
| arcasHLA | 1.3.2 | git | HLA Typing |
|
|
53
|
+
| TIDEpy | 1.3.6 | git | Immune response prediction |
|
|
54
|
+
| centrifuge | 1.0.4 | bioconda | Microbiome |
|
|
@@ -0,0 +1,39 @@
|
|
|
1
|
+
{
|
|
2
|
+
"$schema": "http://json-schema.org/draft-07/schema#",
|
|
3
|
+
"$id": "output_API.schema",
|
|
4
|
+
"title": "Pipeline output_API.JSON schema",
|
|
5
|
+
"type": "object",
|
|
6
|
+
"description": "Schema for pipeline's output_API.JSONs",
|
|
7
|
+
"properties": {
|
|
8
|
+
"metasheet": {
|
|
9
|
+
"type": "string",
|
|
10
|
+
"pattern": ".*\\.csv$"
|
|
11
|
+
},
|
|
12
|
+
"assembly": {
|
|
13
|
+
"type": "string",
|
|
14
|
+
"const": "hg38"
|
|
15
|
+
},
|
|
16
|
+
"ref": {
|
|
17
|
+
"type": "string",
|
|
18
|
+
"pattern": ".*\\.yaml$"
|
|
19
|
+
},
|
|
20
|
+
"mate": {
|
|
21
|
+
"type": "array"
|
|
22
|
+
},
|
|
23
|
+
"rsem": {
|
|
24
|
+
"type": "boolean"
|
|
25
|
+
},
|
|
26
|
+
"stranded": {
|
|
27
|
+
"type": "boolean"
|
|
28
|
+
},
|
|
29
|
+
"samples": {
|
|
30
|
+
"type": "object"
|
|
31
|
+
},
|
|
32
|
+
"runs": {
|
|
33
|
+
"type": "object"
|
|
34
|
+
}
|
|
35
|
+
}
|
|
36
|
+
}
|
|
37
|
+
|
|
38
|
+
|
|
39
|
+
|
|
@@ -0,0 +1,195 @@
|
|
|
1
|
+
{
|
|
2
|
+
"cimac id": [
|
|
3
|
+
{
|
|
4
|
+
"filter_group": "",
|
|
5
|
+
"file_path_template": "analysis/{cimac id}_error.yaml",
|
|
6
|
+
"short_description": "yaml file that specifies error codes for files",
|
|
7
|
+
"long_description": "Explanation of all files which are expected to be empty due to a failed/missing module.",
|
|
8
|
+
"file_purpose": "Analysis view",
|
|
9
|
+
"optional": true
|
|
10
|
+
},
|
|
11
|
+
{
|
|
12
|
+
"filter_group": "alignment",
|
|
13
|
+
"file_path_template": "analysis/star/{cimac id}/{cimac id}.sorted.bam",
|
|
14
|
+
"short_description": "star alignment output",
|
|
15
|
+
"long_description": "Alignments in binary BAM format sorted by coordinate Aligned.",
|
|
16
|
+
"file_purpose": "Analysis view"
|
|
17
|
+
},
|
|
18
|
+
{
|
|
19
|
+
"filter_group": "alignment",
|
|
20
|
+
"file_path_template": "analysis/star/{cimac id}/{cimac id}.sorted.bam.bai",
|
|
21
|
+
"short_description": "sorted_bam_index file",
|
|
22
|
+
"long_description": "file sorted_bam_index file sorted_bam_index file sorted_bam_index file",
|
|
23
|
+
"file_purpose": "Miscellaneous"
|
|
24
|
+
},
|
|
25
|
+
{
|
|
26
|
+
"filter_group": "alignment",
|
|
27
|
+
"file_path_template": "analysis/star/{cimac id}/{cimac id}.sorted.bam.stat.txt",
|
|
28
|
+
"short_description": "sorted_bam_stat_txt file",
|
|
29
|
+
"long_description": "sorted_bam_stat_txt file sorted_bam_stat_txt sorted_bam_stat_txt",
|
|
30
|
+
"file_purpose": "Miscellaneous"
|
|
31
|
+
},
|
|
32
|
+
|
|
33
|
+
{
|
|
34
|
+
"filter_group": "alignment",
|
|
35
|
+
"file_path_template": "analysis/star/{cimac id}/{cimac id}.transcriptome.bam",
|
|
36
|
+
"short_description": "transcriptome bam file",
|
|
37
|
+
"long_description": "file transcriptome_bam file transcriptome_bam file transcriptome_bam file",
|
|
38
|
+
"file_purpose": "Miscellaneous"
|
|
39
|
+
},
|
|
40
|
+
|
|
41
|
+
{
|
|
42
|
+
"filter_group": "alignment",
|
|
43
|
+
"file_path_template": "analysis/star/{cimac id}/{cimac id}.Chimeric.out.junction",
|
|
44
|
+
"short_description": "Chimeric junction output",
|
|
45
|
+
"long_description": "Chimeric junction output for fusion calling",
|
|
46
|
+
"file_purpose": "Miscellaneous"
|
|
47
|
+
},
|
|
48
|
+
|
|
49
|
+
|
|
50
|
+
{
|
|
51
|
+
"filter_group": "quality",
|
|
52
|
+
"file_path_template": "analysis/rseqc/read_distrib/{cimac id}/{cimac id}.txt",
|
|
53
|
+
"short_description": "read distribution ouput",
|
|
54
|
+
"long_description": "file read_distrib file read_distrib file read_distrib file",
|
|
55
|
+
"file_purpose": "Clinical view"
|
|
56
|
+
},
|
|
57
|
+
{
|
|
58
|
+
"filter_group": "quality",
|
|
59
|
+
"file_path_template": "analysis/rseqc/tin_score/{cimac id}/{cimac id}.summary.txt",
|
|
60
|
+
"short_description": "tin_score_summary file",
|
|
61
|
+
"long_description": "file tin_score_summary file tin_score_summary file tin_score_summary file",
|
|
62
|
+
"file_purpose": "Miscellaneous"
|
|
63
|
+
},
|
|
64
|
+
{
|
|
65
|
+
"filter_group": "quality",
|
|
66
|
+
"file_path_template": "analysis/rseqc/tin_score/{cimac id}/{cimac id}.tin_score.txt",
|
|
67
|
+
"short_description": "tin score output",
|
|
68
|
+
"long_description": "file tin_score file tin_score file tin_score file",
|
|
69
|
+
"file_purpose": "Analysis view"
|
|
70
|
+
},
|
|
71
|
+
|
|
72
|
+
|
|
73
|
+
|
|
74
|
+
{
|
|
75
|
+
"filter_group": "gene-quantification",
|
|
76
|
+
"file_path_template": "analysis/salmon/{cimac id}/{cimac id}.quant.sf",
|
|
77
|
+
"short_description": "quant_sf file",
|
|
78
|
+
"long_description": "file quant_sf file quant_sf file quant_sf file",
|
|
79
|
+
"file_purpose": "Miscellaneous"
|
|
80
|
+
},
|
|
81
|
+
{
|
|
82
|
+
"filter_group": "gene-quantification",
|
|
83
|
+
"file_path_template": "analysis/salmon/{cimac id}/{cimac id}.transcriptome.bam.log",
|
|
84
|
+
"short_description": "transcriptome_bam_log file",
|
|
85
|
+
"long_description": "file transcriptome_bam_log file transcriptome_bam_log file transcriptome_bam_log file",
|
|
86
|
+
"file_purpose": "Miscellaneous"
|
|
87
|
+
},
|
|
88
|
+
{
|
|
89
|
+
"filter_group": "gene-quantification",
|
|
90
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/ambig_info.tsv",
|
|
91
|
+
"short_description": "aux_info_ambig_info_tsv file",
|
|
92
|
+
"long_description": "file aux_info_ambig_info_tsv file aux_info_ambig_info_tsv file aux_info_ambig_info_tsv file",
|
|
93
|
+
"file_purpose": "Miscellaneous"
|
|
94
|
+
},
|
|
95
|
+
{
|
|
96
|
+
"filter_group": "gene-quantification",
|
|
97
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/expected_bias.gz",
|
|
98
|
+
"short_description": "aux_info_expected_bias file",
|
|
99
|
+
"long_description": "file aux_info_expected_bias file aux_info_expected_bias file aux_info_expected_bias file",
|
|
100
|
+
"file_purpose": "Miscellaneous"
|
|
101
|
+
},
|
|
102
|
+
{
|
|
103
|
+
"filter_group": "gene-quantification",
|
|
104
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/fld.gz",
|
|
105
|
+
"short_description": "aux_info_fld file",
|
|
106
|
+
"long_description": "Fragment length didstribution file contains an approximation of observed fragment length distribution",
|
|
107
|
+
"file_purpose": "Miscellaneous"
|
|
108
|
+
},
|
|
109
|
+
{
|
|
110
|
+
"filter_group": "gene-quantification",
|
|
111
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/meta_info.json",
|
|
112
|
+
"short_description": "aux_info_meta_info file",
|
|
113
|
+
"long_description": "meta information about the run, including stats such as the number of observed and mapped fragments, details of the bias modeling etc",
|
|
114
|
+
"file_purpose": "Miscellaneous"
|
|
115
|
+
},
|
|
116
|
+
{
|
|
117
|
+
"filter_group": "gene-quantification",
|
|
118
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/observed_bias.gz",
|
|
119
|
+
"short_description": "aux_info_observed_bias file",
|
|
120
|
+
"long_description": "file aux_info_observed_bias file aux_info_observed_bias file aux_info_observed_bias file",
|
|
121
|
+
"file_purpose": "Miscellaneous"
|
|
122
|
+
},
|
|
123
|
+
{
|
|
124
|
+
"filter_group": "gene-quantification",
|
|
125
|
+
"file_path_template": "analysis/salmon/{cimac id}/aux_info/observed_bias_3p.gz",
|
|
126
|
+
"short_description": "aux_info_observed_bias_3p file",
|
|
127
|
+
"long_description": "file aux_info_observed_bias_3p file aux_info_observed_bias_3p file aux_info_observed_bias_3p file",
|
|
128
|
+
"file_purpose": "Miscellaneous"
|
|
129
|
+
},
|
|
130
|
+
{
|
|
131
|
+
"filter_group": "gene-quantification",
|
|
132
|
+
"file_path_template": "analysis/salmon/{cimac id}/cmd_info.json",
|
|
133
|
+
"short_description": "cmd_info file",
|
|
134
|
+
"long_description": "A file that records the main command line parameters with which Salmon used",
|
|
135
|
+
"file_purpose": "Miscellaneous"
|
|
136
|
+
},
|
|
137
|
+
{
|
|
138
|
+
"filter_group": "gene-quantification",
|
|
139
|
+
"file_path_template": "analysis/salmon/{cimac id}/logs/salmon_quant.log",
|
|
140
|
+
"short_description": "salmon_quant_log file",
|
|
141
|
+
"long_description": "file salmon_quant_log file salmon_quant_log file salmon_quant_log file",
|
|
142
|
+
"file_purpose": "Miscellaneous"
|
|
143
|
+
},
|
|
144
|
+
|
|
145
|
+
|
|
146
|
+
|
|
147
|
+
{
|
|
148
|
+
"filter_group": "microbiome",
|
|
149
|
+
"file_path_template": "analysis/microbiome/{cimac id}/{cimac id}_addSample_report.txt",
|
|
150
|
+
"short_description": "Centrifuge summary output file",
|
|
151
|
+
"long_description": "Centrifuge output file contains name of a genome, taxonomic ID and rank, and also the proportion of this genome normalized by its genomic length",
|
|
152
|
+
"file_purpose": "Miscellaneous"
|
|
153
|
+
},
|
|
154
|
+
|
|
155
|
+
|
|
156
|
+
{
|
|
157
|
+
"filter_group": "immune-repertoire",
|
|
158
|
+
"file_path_template": "analysis/trust4/{cimac id}/{cimac id}_report.tsv",
|
|
159
|
+
"short_description": "TURST4 final report file",
|
|
160
|
+
"long_description": "This report file focuses on CDR3 and is compatible with other repertoire analysis tools, such as VDJTools ",
|
|
161
|
+
"file_purpose": "Miscellaneous"
|
|
162
|
+
},
|
|
163
|
+
|
|
164
|
+
|
|
165
|
+
|
|
166
|
+
{
|
|
167
|
+
"filter_group": "fusion",
|
|
168
|
+
"file_path_template": "analysis/fusion/{cimac id}/{cimac id}.fusion_predictions.abridged_addSample.tsv",
|
|
169
|
+
"short_description": "fusion analysis report file",
|
|
170
|
+
"long_description": "this report file contains valiated fusion gene pairs found in all samples including their gene expression",
|
|
171
|
+
"file_purpose": "Miscellaneous"
|
|
172
|
+
},
|
|
173
|
+
|
|
174
|
+
|
|
175
|
+
|
|
176
|
+
{
|
|
177
|
+
"filter_group": "MSI",
|
|
178
|
+
"file_path_template": "analysis/msisensor/single/{cimac id}/{cimac id}_msisensor.txt",
|
|
179
|
+
"short_description": "msisensor report file",
|
|
180
|
+
"long_description": "this report file contains msi score this report file contains msi score",
|
|
181
|
+
"file_purpose": "Miscellaneous"
|
|
182
|
+
},
|
|
183
|
+
|
|
184
|
+
|
|
185
|
+
|
|
186
|
+
{
|
|
187
|
+
"filter_group": "HLA",
|
|
188
|
+
"file_path_template": "analysis/neoantigen/{cimac id}/{cimac id}.genotype.json",
|
|
189
|
+
"short_description": "arcasHLA report file",
|
|
190
|
+
"long_description": "this report file contains MHC class I&II HLA allels",
|
|
191
|
+
"file_purpose": "Miscellaneous"
|
|
192
|
+
}
|
|
193
|
+
|
|
194
|
+
]
|
|
195
|
+
}
|
|
Binary file
|
|
@@ -0,0 +1,101 @@
|
|
|
1
|
+
# TCR-seq CIMAC-CIDC Report Description
|
|
2
|
+
|
|
3
|
+
### Summary
|
|
4
|
+
|
|
5
|
+
An in-browser immune repertoire report, relying on VisualizIRR (Visualized Immune Repertoire Report), is used make immune repertoire analysis results simple to navigate and understand for the end user on their local machine or on a server.
|
|
6
|
+
The report contains analysis on the cohort and sample level in order to inform the end user of CDR3 distribution, top clonotypes, and V/J-gene usage.
|
|
7
|
+
Additional information related to diversity and clonality is also included in an intracohort analysis module and is displayed on a boxplot that may be split by meta-information, when it is available.
|
|
8
|
+
When multiple chains are available in the dataset, they are seperated.
|
|
9
|
+
Information in the report is displayed mostly through interactive plots which may be exported at a higher resolution.
|
|
10
|
+
|
|
11
|
+
### Decompressing the report file
|
|
12
|
+
|
|
13
|
+
The report can be downloaded from the CIDC portal in tar.gz compressed format. On a Mac, one can simply double-click on the <report_name>.tar.gz file to decompress the file. The file can also be decompressed in a terminal window using the bash command tar:
|
|
14
|
+
|
|
15
|
+
```
|
|
16
|
+
tar -xvzf <report_name>tar.gz
|
|
17
|
+
```
|
|
18
|
+
|
|
19
|
+
On a Windows-based system, a third-party software such as Winzip can be used to decompress the file.
|
|
20
|
+
|
|
21
|
+
### Viewing Report on a Local Machine
|
|
22
|
+
|
|
23
|
+
For security purposes, newer browsers can have problems loading local files normally in an HTML page.
|
|
24
|
+
If you can't view the the display locally by simply opening the html in a browser, you can perform the following in your terminal to set up a simple server and view the report:
|
|
25
|
+
```
|
|
26
|
+
cd <VisualizIRR_directory>
|
|
27
|
+
```
|
|
28
|
+
Python 2:
|
|
29
|
+
```
|
|
30
|
+
python -m SimpleHTTPServer & python -m webbrowser -n "http://0.0.0.0:8000"
|
|
31
|
+
```
|
|
32
|
+
Python 3:
|
|
33
|
+
```
|
|
34
|
+
python3 -m http.server & python3 -m webbrowser -n "http://0.0.0.0:8000"
|
|
35
|
+
```
|
|
36
|
+
|
|
37
|
+
The report is viewable at http://0.0.0.0:8000, which should pop up automatically.
|
|
38
|
+
|
|
39
|
+
### TCR-seq Workflow
|
|
40
|
+
|
|
41
|
+
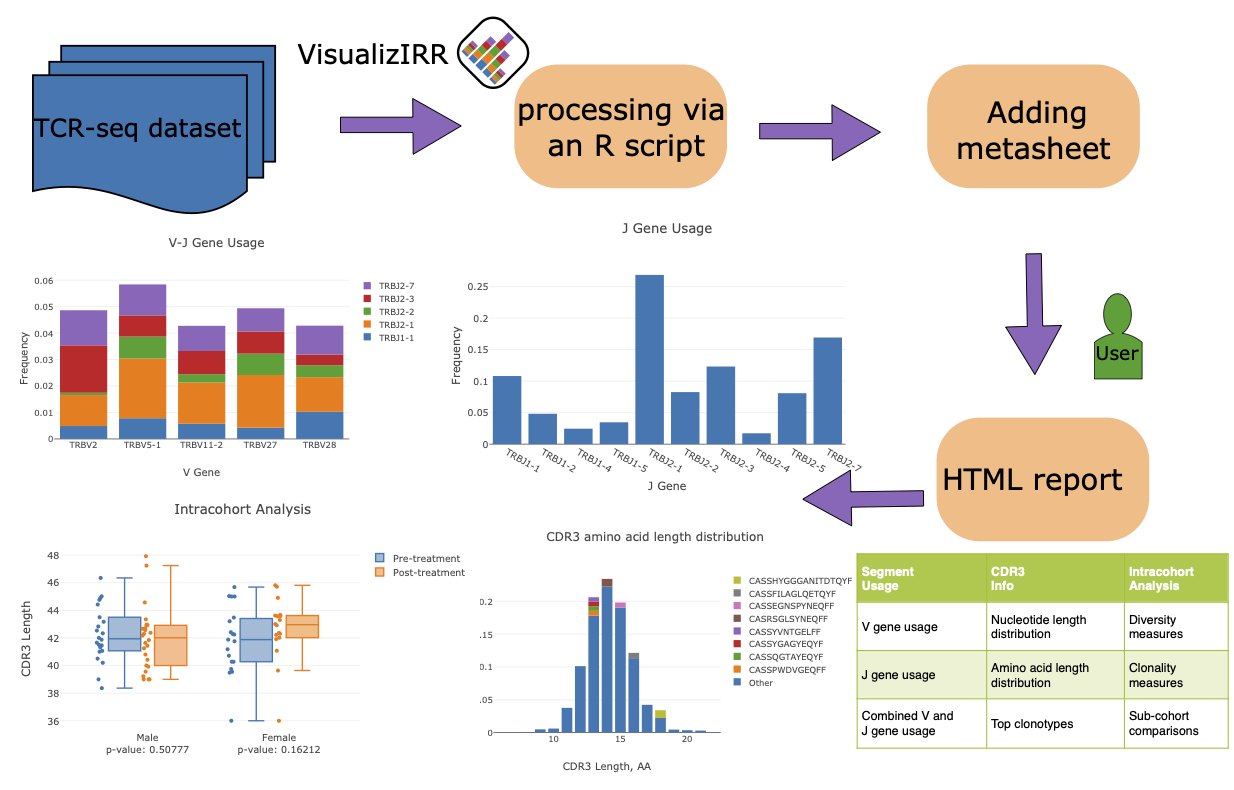
|
|
42
|
+
|
|
43
|
+
### Adding metadata to the report
|
|
44
|
+
**meta.csv** is required for intracohort analysis and is the only component of the final report that needs to be manually composed by the end-user.
|
|
45
|
+
|
|
46
|
+
**meta.csv** can be found within the report folder in the **data** subdirectory.
|
|
47
|
+
|
|
48
|
+
**meta.csv** should be a csv with the first column including sample names and remaining columns for different conditions.
|
|
49
|
+
There are a few ways to enter your meta information.
|
|
50
|
+
|
|
51
|
+
1. In order to have ordered sample condition groups, denote the categorical label of those groups in the header using '|' as the separator and use the corresponding numbers in the sample rows, starting with 0 for the first label (as demonstrated in Timepoint and Condition 1).
|
|
52
|
+
2. You can also use the labels in the metasheet and not denote them in the header (as demonstrated in Age and Condition 2).
|
|
53
|
+
3. Meta-data should be converted to categorical bins if it isn't categorical already (as demonstrated in Age)
|
|
54
|
+
4. In order to set up paired samples analysis, two columns must be in the meta.csv -- 'Timepoint' and 'VisGroup'. 'Timepoint' should contain ordered labels as described above. The column named 'VisGroup' indicates which samples should be paired. (as demonstrated in the meta.csv sample below, SampleName1 and SampleName3 are paired. SampleName1 is a sample taken at Baseline. SampleName3 is a sample taken at Cycle 2.) Therefore, patient samples from different timepoints can be paired.
|
|
55
|
+
|
|
56
|
+
meta.csv template:
|
|
57
|
+
```
|
|
58
|
+
sample,Timepoint|Baseline|Cycle 1|Cycle 2,Age,Condition 1|Group 0|Group 1|Group 2,Condition 2,VisGroup
|
|
59
|
+
SampleName0,1,20-29,0,Aa,0
|
|
60
|
+
SampleName1,0,40-49,2,Bb,1
|
|
61
|
+
SampleName2,1,10-19,1,Cc,2
|
|
62
|
+
SampleName3,2,40-49,2,Bb,1
|
|
63
|
+
```
|
|
64
|
+
|
|
65
|
+
### Video Tutorial
|
|
66
|
+
The following video describes decompressing and opening the report as well as how to modify the meta.csv file.
|
|
67
|
+
|
|
68
|
+
[](https://www.youtube.com/watch?v=vmDwjSrei0c)
|
|
69
|
+
|
|
70
|
+
### Information in Report
|
|
71
|
+
|
|
72
|
+
* Cohort and sample level
|
|
73
|
+
* Segment Usage
|
|
74
|
+
* V gene and J gene usage
|
|
75
|
+
* Combined V and J gene usage
|
|
76
|
+
* CDR3 Info
|
|
77
|
+
* CDR3 length distribution
|
|
78
|
+
* Top clonotypes
|
|
79
|
+
* Intracohort Analysis & Information Table
|
|
80
|
+
* Clonotypes Per Kilo-reads (CPK)
|
|
81
|
+
* Raw Diversity
|
|
82
|
+
* Entropy, 1/Entropy, Normalized Entropy
|
|
83
|
+
* Gini Coefficient
|
|
84
|
+
* Gini-Simpson Index
|
|
85
|
+
* Inverse Simpson Index
|
|
86
|
+
* Chao1 Index
|
|
87
|
+
* Clonal Proportionality Information
|
|
88
|
+
* Average CDR3 Length
|
|
89
|
+
* Unique CDR3 Count
|
|
90
|
+
|
|
91
|
+
### Tools used
|
|
92
|
+
|
|
93
|
+
The cidc version of VisualizIRR uses the following R based tools:
|
|
94
|
+
* R version 3.5.2
|
|
95
|
+
|
|
96
|
+
* data.table 1.12.8
|
|
97
|
+
* immunarch 0.6.5
|
|
98
|
+
* naturalsort 0.1.3
|
|
99
|
+
* dplyr 1.0.0
|
|
100
|
+
* rjson 0.2.20
|
|
101
|
+
|
|
Binary file
|
|
@@ -0,0 +1,46 @@
|
|
|
1
|
+
## Whole-exome sequencing (WES) analysis pipeline description
|
|
2
|
+
|
|
3
|
+
The CIDC whole-exome sequencing (WES) pipeline aims to identify and characterize key immunotherapeutic features of tumor samples. WES implements the [Gene Analysis Toolkit](https://gatk.broadinstitute.org/hc/en-us) (GATK) best practices and identifies both somatic and germline variants using Sentieon's TNScope and Haplotyper algorithms, respectively. Somatic variants are annotated using the Variant Effect Predictor software. As recommended in [Chen YC, Seifuddin F, et al. 2021](https://www.biorxiv.org/content/10.1101/2021.02.18.431906v1.full), the pipeline uses an ensemble of three callers (CNVkit, Sequenza, and Facets) to characterize tumor copy number variation: the overlap of the CNV segments is used to generate a high-confident consensus set. WES estimates tumor purity using two different software packages, Sequenza and FACETS, and also infers tumor clonal populations using PyClone-VI. The pipeline characterizes tumor HLA type (both class I and class II alleles) using HLA-HD, xHLA, and Optitype. WES also performs neoantigen prediction with pVACtools version 2.0.7 which incorporates netMHCpan 4.1 and netMHCpanII 4.0 as neoantigen callers and IEDB 3.1.1 to predict MHC peptide-MHC binding affinities and prioritize candidate epitopes. WES estimates tumor tcell fraction using TCellExTRECT and tumor microsatellite instability using MSIsensor2.
|
|
4
|
+
|
|
5
|
+
### WES Workflow
|
|
6
|
+
|
|
7
|
+
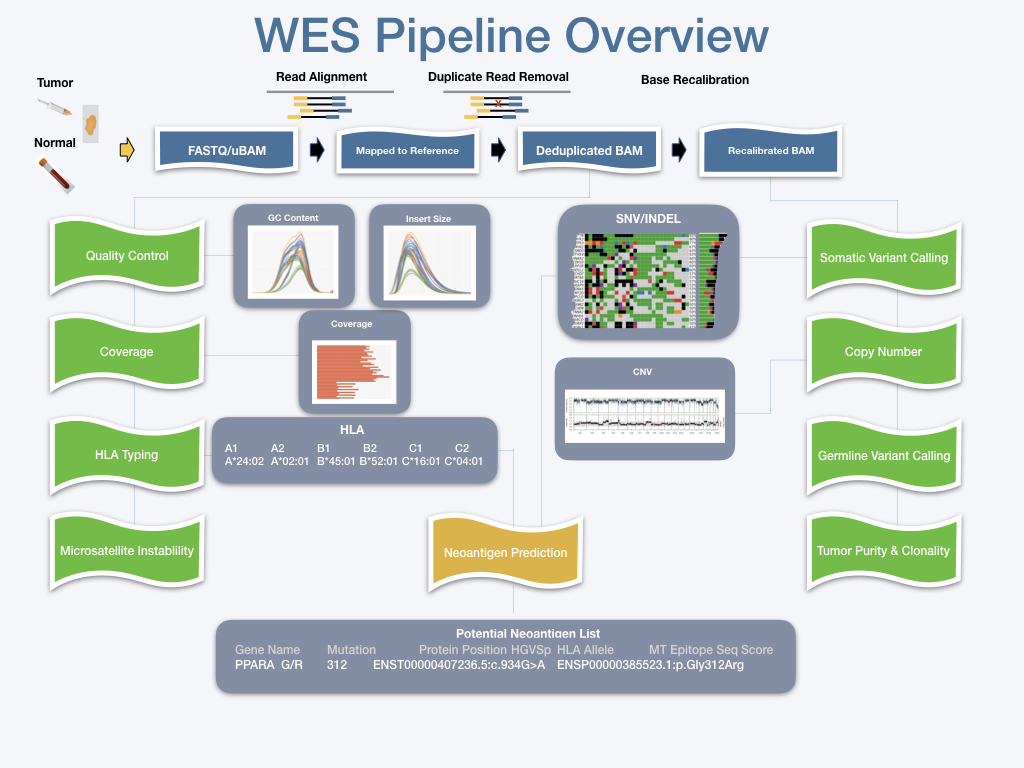
|
|
8
|
+
|
|
9
|
+
|
|
10
|
+
## Versions of Tools and Reference Files Used in WES
|
|
11
|
+
|
|
12
|
+
| Reference/Software | Version | Source | Notes |
|
|
13
|
+
|--|--|--|--|
|
|
14
|
+
| Assembly | hg38 | GDC | modified to only include chr1-22,X,Y,M |
|
|
15
|
+
| Sentieon TNscope | sentieon-genomics-202010.01 | Sentieon | Somatic variant caller |
|
|
16
|
+
| Sentieon Haplotyper | sentieon-genomics-202010.01 | Sentieon | Germline variant caller |
|
|
17
|
+
| VEP | 91.3 | Ensembl | Variant annotation |
|
|
18
|
+
| CNVkit | 0.9.9 | bioconda | Copy number variation |
|
|
19
|
+
| Sequenza | 2.1.2 | biobuilds | Copy number variation; Tumor purity/ploidy |
|
|
20
|
+
| Sequenza-utils | 2.1.9999b0 | pip | Copy number variation; Tumor purity/ploidy |
|
|
21
|
+
| Facets | 0.5.14 | bioconda | Copy number variation; Tumor purity/ploidy |
|
|
22
|
+
| PyClone-VI | 0.1.1 | pip | Tumor clonality |
|
|
23
|
+
| HLA-HD | 1.4.0 | [website](https://www.genome.med.kyoto-u.ac.jp/HLA-HD/) | HLA typing |
|
|
24
|
+
| xHLA | commit 34221ea | [github](https://github.com/humanlongevity/HLA) | HLA typing |
|
|
25
|
+
| pVACtools | 2.0.7 | pip | Neoantigen prediction |
|
|
26
|
+
| IEDB | 3.1.1 | [website](https://downloads.iedb.org/tools/mhci/3.1.1/)| Epitope database |
|
|
27
|
+
| TcellExTRECT | commit ec81143 | [github](https://github.com/McGranahanLab/TcellExTRECT)| Epitope database |
|
|
28
|
+
| MSIsensor2 | v0.1 | [github](https://github.com/niu-lab/msisensor2.git) | Microsatellite instability |
|
|
29
|
+
|
|
30
|
+
---
|
|
31
|
+
## ExACdb assembly compatibility issue
|
|
32
|
+
|
|
33
|
+
### Issue:
|
|
34
|
+
The version of vcf2maf (v1.6.18) included in the CIDC WES pipeline uses ExACdb. CIDC WES uses the hg38 gene model. ExACdb is based on a hg19 reference, not hg38. Therefore some of the CIDC WES variants annotated by vcf2maf using ExACdb might not be accurate as the hg19/hg38 variant locations might be discrepant.
|
|
35
|
+
|
|
36
|
+
### CIDC Portal Files Affected (WES only):
|
|
37
|
+
The columns listed below in the output.twist.maf and output.twist.filtered.maf files.
|
|
38
|
+
|
|
39
|
+
### MAF fields affected in these files:
|
|
40
|
+
FILTER (though original FILTER flag is preserved; the ExAC_FILTER is added)
|
|
41
|
+
|
|
42
|
+
ExAC_FILTER, ExAC_AF_Adj, ExAC_AC_AN_Adj, ExAC_AC_AN, ExAC_AC_AN_AFR, ExAC_AC_AN_AMR, ExAC_AC_AN_EAS, ExAC_AC_AN_FIN, ExAC_AC_AN_NFE, ExAC_AC_AN_OTH, ExAC_AC_AN_SAS
|
|
43
|
+
|
|
44
|
+
Please see this [link](https://github.com/mskcc/vcf2maf/blob/47c4a18a15d5f93d4f3622615b3368448a74127d/docs/vep_maf_readme.txt) for more information about these fields.
|
|
45
|
+
|
|
46
|
+
---
|