sortscore 0.1.0__tar.gz
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- sortscore-0.1.0/LICENSE +21 -0
- sortscore-0.1.0/PKG-INFO +75 -0
- sortscore-0.1.0/README.md +47 -0
- sortscore-0.1.0/pyproject.toml +43 -0
- sortscore-0.1.0/setup.cfg +4 -0
- sortscore-0.1.0/sortscore/__init__.py +19 -0
- sortscore-0.1.0/sortscore/__main__.py +7 -0
- sortscore-0.1.0/sortscore/analysis/__init__.py +7 -0
- sortscore-0.1.0/sortscore/analysis/aa_scores.py +214 -0
- sortscore-0.1.0/sortscore/analysis/annotation.py +188 -0
- sortscore-0.1.0/sortscore/analysis/batch_config.py +170 -0
- sortscore-0.1.0/sortscore/analysis/batch_normalization.py +978 -0
- sortscore-0.1.0/sortscore/analysis/batch_workflow.py +70 -0
- sortscore-0.1.0/sortscore/analysis/filtering.py +53 -0
- sortscore-0.1.0/sortscore/analysis/normalize_read_depth.py +99 -0
- sortscore-0.1.0/sortscore/analysis/score.py +243 -0
- sortscore-0.1.0/sortscore/analysis/statistics.py +197 -0
- sortscore-0.1.0/sortscore/analysis/summary_stats.py +161 -0
- sortscore-0.1.0/sortscore/analysis/variant_aggregation.py +94 -0
- sortscore-0.1.0/sortscore/analysis/workflows.py +231 -0
- sortscore-0.1.0/sortscore/cli.py +48 -0
- sortscore-0.1.0/sortscore/run_analysis.py +221 -0
- sortscore-0.1.0/sortscore/run_batch_analysis.py +81 -0
- sortscore-0.1.0/sortscore/utils/analysis_logger.py +268 -0
- sortscore-0.1.0/sortscore/utils/console_utils.py +203 -0
- sortscore-0.1.0/sortscore/utils/experiment_setup.py +325 -0
- sortscore-0.1.0/sortscore/utils/file_utils.py +158 -0
- sortscore-0.1.0/sortscore/utils/load_experiment.py +675 -0
- sortscore-0.1.0/sortscore/utils/sequence_parsing.py +318 -0
- sortscore-0.1.0/sortscore/utils/tile_configs.py +115 -0
- sortscore-0.1.0/sortscore/utils/variant_detection.py +329 -0
- sortscore-0.1.0/sortscore/utils/variant_parsing.py +68 -0
- sortscore-0.1.0/sortscore/visualization/__init__.py +5 -0
- sortscore-0.1.0/sortscore/visualization/correlations.py +358 -0
- sortscore-0.1.0/sortscore/visualization/heatmap_matrix.py +180 -0
- sortscore-0.1.0/sortscore/visualization/heatmap_workflow.py +213 -0
- sortscore-0.1.0/sortscore/visualization/heatmaps.py +728 -0
- sortscore-0.1.0/sortscore/visualization/plots.py +358 -0
- sortscore-0.1.0/sortscore.egg-info/PKG-INFO +75 -0
- sortscore-0.1.0/sortscore.egg-info/SOURCES.txt +42 -0
- sortscore-0.1.0/sortscore.egg-info/dependency_links.txt +1 -0
- sortscore-0.1.0/sortscore.egg-info/entry_points.txt +2 -0
- sortscore-0.1.0/sortscore.egg-info/requires.txt +7 -0
- sortscore-0.1.0/sortscore.egg-info/top_level.txt +1 -0
sortscore-0.1.0/LICENSE
ADDED
|
@@ -0,0 +1,21 @@
|
|
|
1
|
+
MIT License
|
|
2
|
+
|
|
3
|
+
Copyright (c) 2025 dbaldridge-lab
|
|
4
|
+
|
|
5
|
+
Permission is hereby granted, free of charge, to any person obtaining a copy
|
|
6
|
+
of this software and associated documentation files (the "Software"), to deal
|
|
7
|
+
in the Software without restriction, including without limitation the rights
|
|
8
|
+
to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
|
|
9
|
+
copies of the Software, and to permit persons to whom the Software is
|
|
10
|
+
furnished to do so, subject to the following conditions:
|
|
11
|
+
|
|
12
|
+
The above copyright notice and this permission notice shall be included in all
|
|
13
|
+
copies or substantial portions of the Software.
|
|
14
|
+
|
|
15
|
+
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR
|
|
16
|
+
IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
|
|
17
|
+
FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE
|
|
18
|
+
AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER
|
|
19
|
+
LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM,
|
|
20
|
+
OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE
|
|
21
|
+
SOFTWARE.
|
sortscore-0.1.0/PKG-INFO
ADDED
|
@@ -0,0 +1,75 @@
|
|
|
1
|
+
Metadata-Version: 2.4
|
|
2
|
+
Name: sortscore
|
|
3
|
+
Version: 0.1.0
|
|
4
|
+
Summary: A modular Python package for Sort-seq variant analysis
|
|
5
|
+
Author-email: Caitlyn Chitwood <c.chitwood@wustl.edu>
|
|
6
|
+
License: MIT
|
|
7
|
+
Project-URL: Homepage, https://github.com/dbaldridge-lab/sortscore
|
|
8
|
+
Keywords: bioinformatics,sequencing,variant-analysis,sort-seq
|
|
9
|
+
Classifier: Development Status :: 5 - Production/Stable
|
|
10
|
+
Classifier: Intended Audience :: Science/Research
|
|
11
|
+
Classifier: Topic :: Scientific/Engineering :: Bio-Informatics
|
|
12
|
+
Classifier: License :: OSI Approved :: MIT License
|
|
13
|
+
Classifier: Programming Language :: Python :: 3
|
|
14
|
+
Classifier: Programming Language :: Python :: 3.10
|
|
15
|
+
Classifier: Programming Language :: Python :: 3.11
|
|
16
|
+
Classifier: Programming Language :: Python :: 3.12
|
|
17
|
+
Requires-Python: >=3.10
|
|
18
|
+
Description-Content-Type: text/markdown
|
|
19
|
+
License-File: LICENSE
|
|
20
|
+
Requires-Dist: pandas>=2.0.0
|
|
21
|
+
Requires-Dist: numpy>=1.24.0
|
|
22
|
+
Requires-Dist: scipy>=1.10.0
|
|
23
|
+
Requires-Dist: matplotlib>=3.6.0
|
|
24
|
+
Requires-Dist: seaborn>=0.12.0
|
|
25
|
+
Requires-Dist: biopython>=1.81
|
|
26
|
+
Requires-Dist: mavehgvs>=0.7.0
|
|
27
|
+
Dynamic: license-file
|
|
28
|
+
|
|
29
|
+
# sortscore
|
|
30
|
+
|
|
31
|
+
`sortscore` is a Python package for Sort-seq variant analysis, including scoring, normalization, and visualization.
|
|
32
|
+
|
|
33
|
+
## Quick Start
|
|
34
|
+
|
|
35
|
+
```bash
|
|
36
|
+
python -m venv .venv
|
|
37
|
+
source .venv/bin/activate
|
|
38
|
+
pip install sortscore
|
|
39
|
+
```
|
|
40
|
+
Run a standard variant scoring analysis:
|
|
41
|
+
|
|
42
|
+
```bash
|
|
43
|
+
sortscore -n EXPERIMENT_NAME -e path/to/experiment_setup.csv -c path/to/config.json
|
|
44
|
+
```
|
|
45
|
+
If the `sortscore` command is not on your `PATH`, run:
|
|
46
|
+
|
|
47
|
+
```bash
|
|
48
|
+
python -m sortscore -n EXPERIMENT_NAME -e path/to/experiment_setup.csv -c path/to/config.json
|
|
49
|
+
```
|
|
50
|
+
|
|
51
|
+
## Example Heatmaps
|
|
52
|
+
|
|
53
|
+
Single Experiment Scoring Heatmap
|
|
54
|
+
|
|
55
|
+
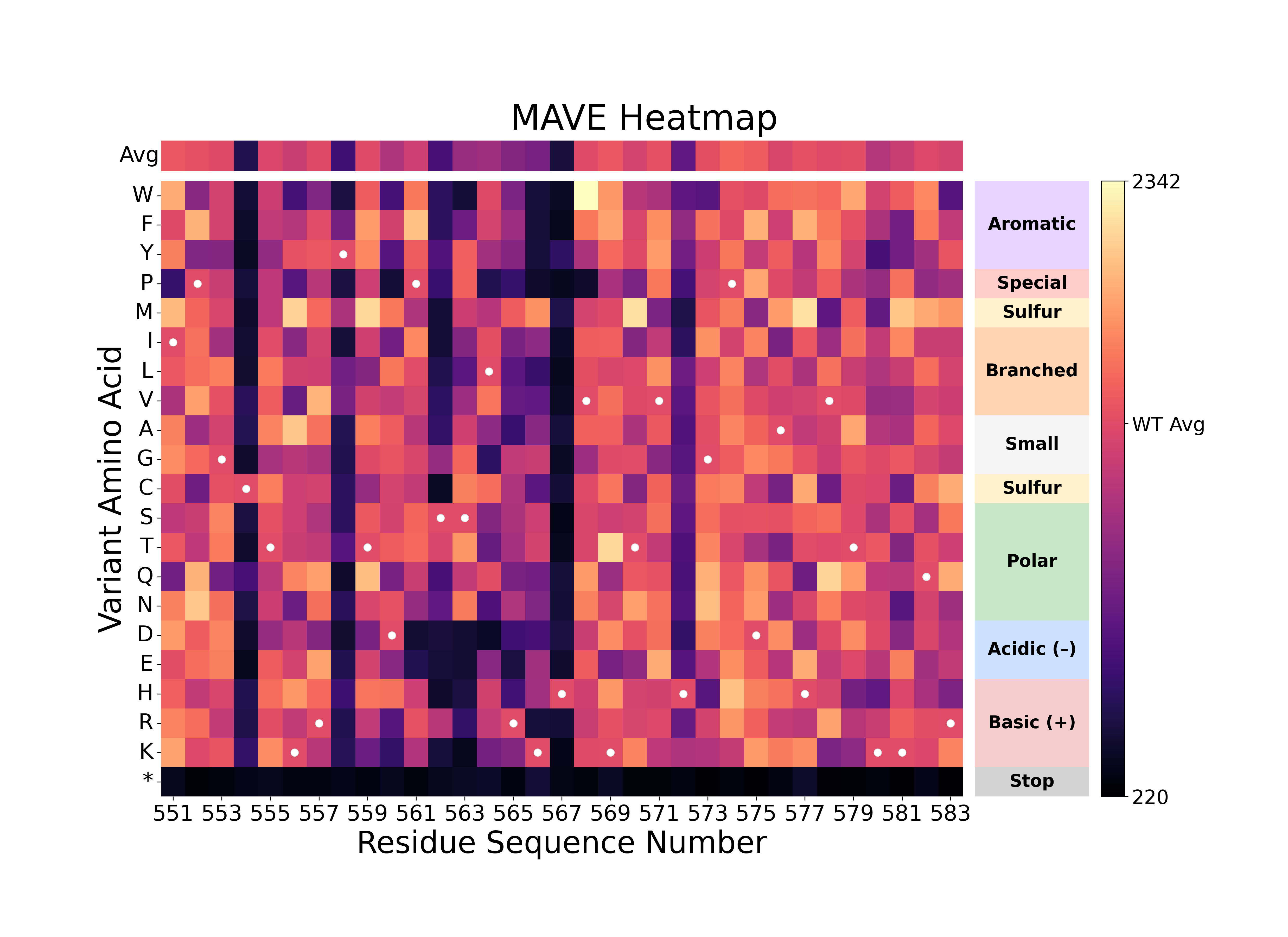
|
|
56
|
+
|
|
57
|
+
Batch Normalization Heatmap
|
|
58
|
+
|
|
59
|
+
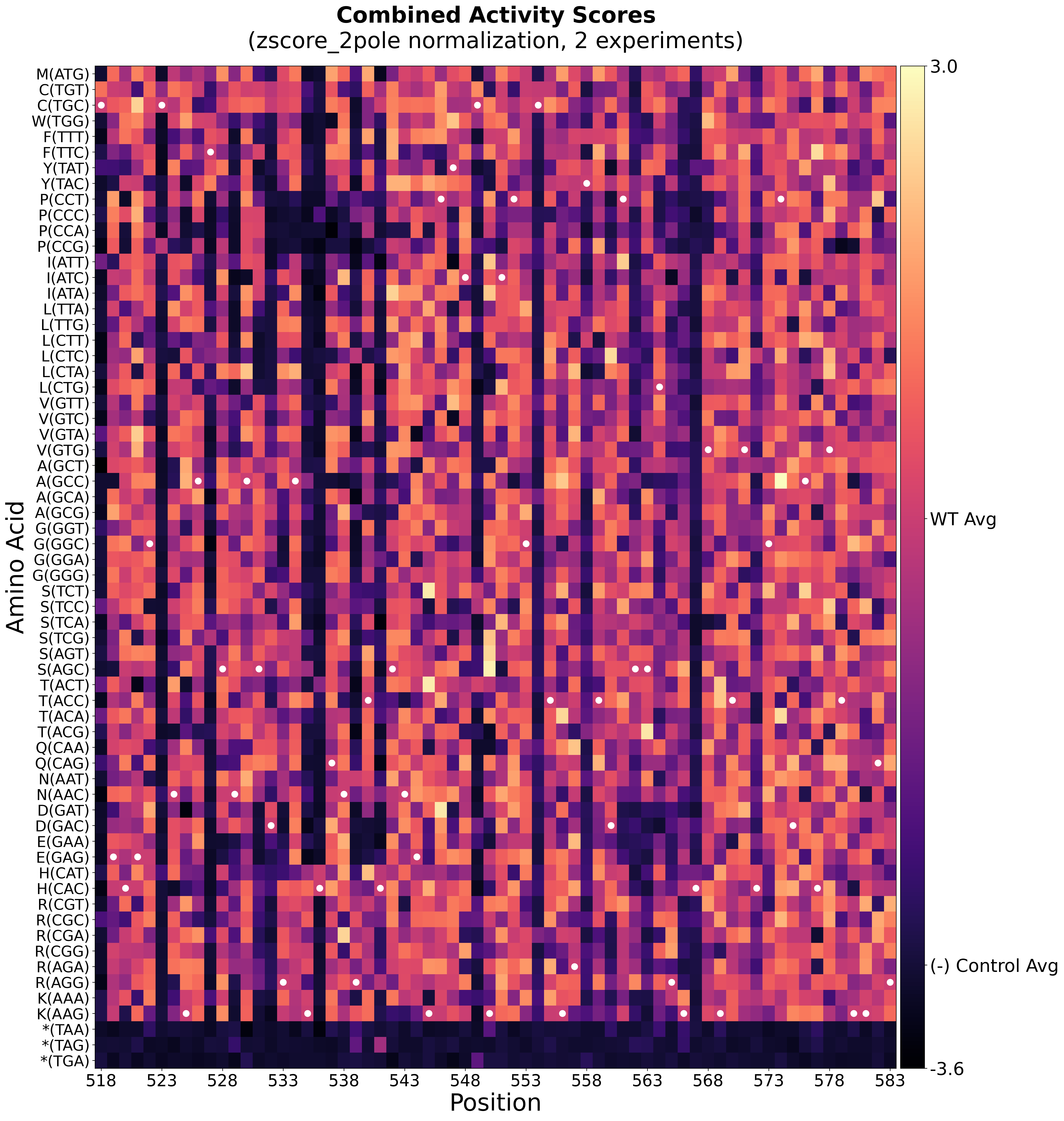
|
|
60
|
+
|
|
61
|
+
## Documentation
|
|
62
|
+
- [Installation](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/installation.md)
|
|
63
|
+
- [Usage](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/usage.md)
|
|
64
|
+
- [CLI Arguments](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/cli_arguments.md)
|
|
65
|
+
- [Visualization](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/visualization.md)
|
|
66
|
+
- [Batch Processing](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/batch_processing.md)
|
|
67
|
+
- [Troubleshooting](https://github.com/dbaldridge-lab/sortscore/blob/main/TROUBLESHOOTING.md)
|
|
68
|
+
- [Contributing](https://github.com/dbaldridge-lab/sortscore/blob/main/CONTRIBUTING.md)
|
|
69
|
+
|
|
70
|
+
## Demo
|
|
71
|
+
|
|
72
|
+
- [Single Experiment Scoring Notebook Demo](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/single_experiment_demo.ipynb)
|
|
73
|
+
- [Batch Normalization Notebook Demo](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/tiled_demo.ipynb)
|
|
74
|
+
- [Example Config](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/GLI2_oPool5b/config.json)
|
|
75
|
+
- [Example Experiment Setup](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/GLI2_oPool5b/experiment_setup.csv)
|
|
@@ -0,0 +1,47 @@
|
|
|
1
|
+
# sortscore
|
|
2
|
+
|
|
3
|
+
`sortscore` is a Python package for Sort-seq variant analysis, including scoring, normalization, and visualization.
|
|
4
|
+
|
|
5
|
+
## Quick Start
|
|
6
|
+
|
|
7
|
+
```bash
|
|
8
|
+
python -m venv .venv
|
|
9
|
+
source .venv/bin/activate
|
|
10
|
+
pip install sortscore
|
|
11
|
+
```
|
|
12
|
+
Run a standard variant scoring analysis:
|
|
13
|
+
|
|
14
|
+
```bash
|
|
15
|
+
sortscore -n EXPERIMENT_NAME -e path/to/experiment_setup.csv -c path/to/config.json
|
|
16
|
+
```
|
|
17
|
+
If the `sortscore` command is not on your `PATH`, run:
|
|
18
|
+
|
|
19
|
+
```bash
|
|
20
|
+
python -m sortscore -n EXPERIMENT_NAME -e path/to/experiment_setup.csv -c path/to/config.json
|
|
21
|
+
```
|
|
22
|
+
|
|
23
|
+
## Example Heatmaps
|
|
24
|
+
|
|
25
|
+
Single Experiment Scoring Heatmap
|
|
26
|
+
|
|
27
|
+

|
|
28
|
+
|
|
29
|
+
Batch Normalization Heatmap
|
|
30
|
+
|
|
31
|
+

|
|
32
|
+
|
|
33
|
+
## Documentation
|
|
34
|
+
- [Installation](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/installation.md)
|
|
35
|
+
- [Usage](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/usage.md)
|
|
36
|
+
- [CLI Arguments](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/cli_arguments.md)
|
|
37
|
+
- [Visualization](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/visualization.md)
|
|
38
|
+
- [Batch Processing](https://github.com/dbaldridge-lab/sortscore/blob/main/docs/batch_processing.md)
|
|
39
|
+
- [Troubleshooting](https://github.com/dbaldridge-lab/sortscore/blob/main/TROUBLESHOOTING.md)
|
|
40
|
+
- [Contributing](https://github.com/dbaldridge-lab/sortscore/blob/main/CONTRIBUTING.md)
|
|
41
|
+
|
|
42
|
+
## Demo
|
|
43
|
+
|
|
44
|
+
- [Single Experiment Scoring Notebook Demo](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/single_experiment_demo.ipynb)
|
|
45
|
+
- [Batch Normalization Notebook Demo](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/tiled_demo.ipynb)
|
|
46
|
+
- [Example Config](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/GLI2_oPool5b/config.json)
|
|
47
|
+
- [Example Experiment Setup](https://github.com/dbaldridge-lab/sortscore/blob/main/demo_data/GLI2_oPool5b/experiment_setup.csv)
|
|
@@ -0,0 +1,43 @@
|
|
|
1
|
+
[build-system]
|
|
2
|
+
requires = ["setuptools>=68", "wheel"]
|
|
3
|
+
build-backend = "setuptools.build_meta"
|
|
4
|
+
|
|
5
|
+
[project]
|
|
6
|
+
name = "sortscore"
|
|
7
|
+
version = "0.1.0"
|
|
8
|
+
description = "A modular Python package for Sort-seq variant analysis"
|
|
9
|
+
readme = "README.md"
|
|
10
|
+
requires-python = ">=3.10"
|
|
11
|
+
license = { text = "MIT" }
|
|
12
|
+
authors = [
|
|
13
|
+
{ name = "Caitlyn Chitwood", email = "c.chitwood@wustl.edu" }
|
|
14
|
+
]
|
|
15
|
+
keywords = ["bioinformatics", "sequencing", "variant-analysis", "sort-seq"]
|
|
16
|
+
classifiers = [
|
|
17
|
+
"Development Status :: 5 - Production/Stable",
|
|
18
|
+
"Intended Audience :: Science/Research",
|
|
19
|
+
"Topic :: Scientific/Engineering :: Bio-Informatics",
|
|
20
|
+
"License :: OSI Approved :: MIT License",
|
|
21
|
+
"Programming Language :: Python :: 3",
|
|
22
|
+
"Programming Language :: Python :: 3.10",
|
|
23
|
+
"Programming Language :: Python :: 3.11",
|
|
24
|
+
"Programming Language :: Python :: 3.12"
|
|
25
|
+
]
|
|
26
|
+
dependencies = [
|
|
27
|
+
"pandas>=2.0.0",
|
|
28
|
+
"numpy>=1.24.0",
|
|
29
|
+
"scipy>=1.10.0",
|
|
30
|
+
"matplotlib>=3.6.0",
|
|
31
|
+
"seaborn>=0.12.0",
|
|
32
|
+
"biopython>=1.81",
|
|
33
|
+
"mavehgvs>=0.7.0"
|
|
34
|
+
]
|
|
35
|
+
|
|
36
|
+
[project.urls]
|
|
37
|
+
Homepage = "https://github.com/dbaldridge-lab/sortscore"
|
|
38
|
+
|
|
39
|
+
[project.scripts]
|
|
40
|
+
sortscore = "sortscore.cli:main"
|
|
41
|
+
|
|
42
|
+
[tool.setuptools.packages.find]
|
|
43
|
+
include = ["sortscore", "sortscore.*"]
|
|
@@ -0,0 +1,19 @@
|
|
|
1
|
+
"""
|
|
2
|
+
sortscore: A modular Python package for Sort-seq variant analysis.
|
|
3
|
+
|
|
4
|
+
This package provides tools for analyzing Sort-seq experimental data,
|
|
5
|
+
calculating activity scores, and generating visualizations.
|
|
6
|
+
"""
|
|
7
|
+
|
|
8
|
+
__version__ = "0.1.0"
|
|
9
|
+
__author__ = "Caitlyn Chitwood"
|
|
10
|
+
__email__ = "c.chitwood@wustl.edu"
|
|
11
|
+
|
|
12
|
+
# Import main classes for convenience
|
|
13
|
+
from .utils.load_experiment import ExperimentConfig
|
|
14
|
+
from .analysis.score import calculate_full_activity_scores
|
|
15
|
+
|
|
16
|
+
__all__ = [
|
|
17
|
+
"ExperimentConfig",
|
|
18
|
+
"calculate_full_activity_scores"
|
|
19
|
+
]
|
|
@@ -0,0 +1,214 @@
|
|
|
1
|
+
"""
|
|
2
|
+
Amino acid scores processing and export for Sort-seq analysis.
|
|
3
|
+
|
|
4
|
+
This module provides functions for processing amino acid scores from DNA variant data,
|
|
5
|
+
including aggregation of synonymous codons, statistical analysis, and file export.
|
|
6
|
+
"""
|
|
7
|
+
import os
|
|
8
|
+
import logging
|
|
9
|
+
import pandas as pd
|
|
10
|
+
import numpy as np
|
|
11
|
+
from scipy import stats as scipy_stats
|
|
12
|
+
from typing import List
|
|
13
|
+
from sortscore.analysis.statistics import calculate_codon_and_replicate_variance
|
|
14
|
+
|
|
15
|
+
|
|
16
|
+
def _get_score_column_from_avg_method(avg_method: str) -> str:
|
|
17
|
+
"""Return the score column name for a given averaging method."""
|
|
18
|
+
if avg_method == 'simple-avg':
|
|
19
|
+
return 'avgscore'
|
|
20
|
+
return f"avgscore_{avg_method.replace('-', '_')}"
|
|
21
|
+
|
|
22
|
+
|
|
23
|
+
def process_and_save_aa_scores(scores_df: pd.DataFrame, experiment, scores_dir: str, analysis_logger) -> None:
|
|
24
|
+
"""
|
|
25
|
+
Process and save amino acid scores from variant data.
|
|
26
|
+
|
|
27
|
+
This function handles the complete AA scores workflow including:
|
|
28
|
+
- Filtering out NaN values
|
|
29
|
+
- Checking if codon aggregation is needed
|
|
30
|
+
- Calculating appropriate statistics (with/without codon variance)
|
|
31
|
+
- Rounding score columns
|
|
32
|
+
- Saving to CSV file
|
|
33
|
+
- Logging output
|
|
34
|
+
|
|
35
|
+
Parameters
|
|
36
|
+
----------
|
|
37
|
+
scores_df : pd.DataFrame
|
|
38
|
+
DataFrame containing variant scores and annotations
|
|
39
|
+
experiment : ExperimentConfig
|
|
40
|
+
Experiment configuration containing metadata
|
|
41
|
+
scores_dir : str
|
|
42
|
+
Directory to save scores file
|
|
43
|
+
analysis_logger : AnalysisLogger
|
|
44
|
+
Logger instance for recording outputs
|
|
45
|
+
|
|
46
|
+
Examples
|
|
47
|
+
--------
|
|
48
|
+
>>> process_and_save_aa_scores(scores_df, experiment, 'output/scores', 'suffix', logger)
|
|
49
|
+
"""
|
|
50
|
+
if 'aa_seq_diff' not in scores_df.columns:
|
|
51
|
+
return
|
|
52
|
+
|
|
53
|
+
score_col = _get_score_column_from_avg_method(experiment.avg_method)
|
|
54
|
+
|
|
55
|
+
aa_scores = build_aa_scores_table(scores_df, score_col)
|
|
56
|
+
|
|
57
|
+
# Save to file
|
|
58
|
+
aa_scores_file = os.path.join(scores_dir, f"{experiment.experiment_name}_aa_scores.csv")
|
|
59
|
+
aa_scores.to_csv(aa_scores_file, index=False)
|
|
60
|
+
logging.info(f"Saved AA scores to {aa_scores_file} ({len(aa_scores)} unique AA variants)")
|
|
61
|
+
|
|
62
|
+
# Log file output
|
|
63
|
+
analysis_logger.log_output_file(
|
|
64
|
+
'aa_scores',
|
|
65
|
+
f"{experiment.experiment_name}_aa_scores.csv",
|
|
66
|
+
aa_scores_file,
|
|
67
|
+
variant_count=len(aa_scores)
|
|
68
|
+
)
|
|
69
|
+
|
|
70
|
+
|
|
71
|
+
def build_aa_scores_table(scores_df: pd.DataFrame, score_col: str) -> pd.DataFrame:
|
|
72
|
+
"""
|
|
73
|
+
Build the AA-level score table from a scored variant table.
|
|
74
|
+
|
|
75
|
+
Parameters
|
|
76
|
+
----------
|
|
77
|
+
scores_df : pd.DataFrame
|
|
78
|
+
DataFrame containing variant scores and annotations.
|
|
79
|
+
score_col : str
|
|
80
|
+
Score column used to determine which rows are retained and how codon
|
|
81
|
+
variance is calculated.
|
|
82
|
+
|
|
83
|
+
Returns
|
|
84
|
+
-------
|
|
85
|
+
pd.DataFrame
|
|
86
|
+
AA-level score table with synonymous codons aggregated when needed.
|
|
87
|
+
"""
|
|
88
|
+
if 'aa_seq_diff' not in scores_df.columns:
|
|
89
|
+
raise ValueError("scores_df must contain 'aa_seq_diff' column for AA score output")
|
|
90
|
+
|
|
91
|
+
scores_df_drop_nan = scores_df.dropna(subset=[score_col])
|
|
92
|
+
rep_score_columns = [
|
|
93
|
+
col for col in scores_df_drop_nan.columns
|
|
94
|
+
if col.startswith('Rep') and col.endswith('.score')
|
|
95
|
+
]
|
|
96
|
+
return _check_codon_num(scores_df_drop_nan, score_col, rep_score_columns)
|
|
97
|
+
|
|
98
|
+
|
|
99
|
+
def _check_codon_num(scores_df_drop_nan: pd.DataFrame, score_col: str,
|
|
100
|
+
rep_score_columns: List[str]) -> pd.DataFrame:
|
|
101
|
+
"""
|
|
102
|
+
Check if codon aggregation is needed and process AA scores accordingly.
|
|
103
|
+
|
|
104
|
+
This function checks if there are multiple codons per AA variant and processes
|
|
105
|
+
the data using either DNA->AA aggregation (with codon variance) or AA-only
|
|
106
|
+
statistics (replicate variance only).
|
|
107
|
+
|
|
108
|
+
Parameters
|
|
109
|
+
----------
|
|
110
|
+
scores_df_drop_nan : pd.DataFrame
|
|
111
|
+
DataFrame with NaN values already filtered out
|
|
112
|
+
score_col : str
|
|
113
|
+
Name of the score column to use
|
|
114
|
+
rep_score_columns : List[str]
|
|
115
|
+
List of replicate score column names
|
|
116
|
+
|
|
117
|
+
Returns
|
|
118
|
+
-------
|
|
119
|
+
pd.DataFrame
|
|
120
|
+
Processed AA scores with appropriate statistics
|
|
121
|
+
"""
|
|
122
|
+
# Check if there are multiple codons per AA variant (DNA->AA case)
|
|
123
|
+
aa_variant_counts = scores_df_drop_nan.groupby(['aa_seq_diff', 'annotate_aa']).size()
|
|
124
|
+
needs_aggregation = (aa_variant_counts > 1).any()
|
|
125
|
+
|
|
126
|
+
if needs_aggregation:
|
|
127
|
+
return _process_dna_to_aa_aggregation(scores_df_drop_nan, score_col, rep_score_columns)
|
|
128
|
+
else:
|
|
129
|
+
return _process_aa_only_scores(scores_df_drop_nan, rep_score_columns)
|
|
130
|
+
|
|
131
|
+
|
|
132
|
+
def _process_dna_to_aa_aggregation(scores_df_drop_nan: pd.DataFrame, score_col: str,
|
|
133
|
+
rep_score_columns: List[str]) -> pd.DataFrame:
|
|
134
|
+
"""
|
|
135
|
+
Process DNA->AA aggregation case with codon variance decomposition.
|
|
136
|
+
|
|
137
|
+
Parameters
|
|
138
|
+
----------
|
|
139
|
+
scores_df_drop_nan : pd.DataFrame
|
|
140
|
+
DataFrame with variant scores (NaN filtered)
|
|
141
|
+
score_col : str
|
|
142
|
+
Name of the score column to use
|
|
143
|
+
rep_score_columns : List[str]
|
|
144
|
+
List of replicate score column names
|
|
145
|
+
|
|
146
|
+
Returns
|
|
147
|
+
-------
|
|
148
|
+
pd.DataFrame
|
|
149
|
+
Aggregated AA scores with codon and replicate statistics
|
|
150
|
+
"""
|
|
151
|
+
# DNA->AA aggregation case: aggregate synonymous variants
|
|
152
|
+
columns_to_average = ['avgscore', 'avgscore_rep_weighted'] + rep_score_columns
|
|
153
|
+
|
|
154
|
+
# Calculate standard deviation and count of codon-level scores before AA aggregation
|
|
155
|
+
aa_scores_std = scores_df_drop_nan.groupby(['aa_seq_diff', 'annotate_aa'])[score_col].agg(['std', 'count']).reset_index()
|
|
156
|
+
aa_scores_std.columns = ['aa_seq_diff', 'annotate_aa', 'SD_codon', 'n_codons']
|
|
157
|
+
|
|
158
|
+
# Calculate mean scores for aggregation
|
|
159
|
+
aa_scores = scores_df_drop_nan.groupby(['aa_seq_diff', 'annotate_aa'])[columns_to_average].mean().reset_index()
|
|
160
|
+
|
|
161
|
+
# Merge the standard deviation and count of codon scores
|
|
162
|
+
aa_scores = aa_scores.merge(aa_scores_std, on=['aa_seq_diff', 'annotate_aa'], how='left')
|
|
163
|
+
|
|
164
|
+
# Calculate statistics with codon and replicate variance decomposition
|
|
165
|
+
aa_scores = calculate_codon_and_replicate_variance(aa_scores, rep_score_columns)
|
|
166
|
+
|
|
167
|
+
return aa_scores
|
|
168
|
+
|
|
169
|
+
|
|
170
|
+
def _process_aa_only_scores(scores_df_drop_nan: pd.DataFrame, rep_score_columns: List[str]) -> pd.DataFrame:
|
|
171
|
+
"""
|
|
172
|
+
Process AA-only case with simple replicate statistics.
|
|
173
|
+
|
|
174
|
+
Parameters
|
|
175
|
+
----------
|
|
176
|
+
scores_df_drop_nan : pd.DataFrame
|
|
177
|
+
DataFrame with variant scores (NaN filtered)
|
|
178
|
+
rep_score_columns : List[str]
|
|
179
|
+
List of replicate score column names
|
|
180
|
+
|
|
181
|
+
Returns
|
|
182
|
+
-------
|
|
183
|
+
pd.DataFrame
|
|
184
|
+
AA scores with replicate statistics only
|
|
185
|
+
"""
|
|
186
|
+
# AA-only case: no aggregation needed, just copy the data
|
|
187
|
+
columns_to_include = ['aa_seq_diff', 'annotate_aa', 'avgscore', 'avgscore_rep_weighted'] + rep_score_columns
|
|
188
|
+
|
|
189
|
+
aa_scores = scores_df_drop_nan[columns_to_include].copy()
|
|
190
|
+
|
|
191
|
+
# Calculate simple replicate statistics (no codon variance)
|
|
192
|
+
if len(rep_score_columns) >= 2:
|
|
193
|
+
aa_rep_mean = aa_scores[rep_score_columns].mean(axis=1)
|
|
194
|
+
aa_rep_std = aa_scores[rep_score_columns].std(axis=1, ddof=1)
|
|
195
|
+
|
|
196
|
+
# Calculate n_measurements (just number of non-empty replicates)
|
|
197
|
+
n_measurements = aa_scores[rep_score_columns].notna().sum(axis=1)
|
|
198
|
+
|
|
199
|
+
# Calculate SEM using only replicate variance
|
|
200
|
+
sem = aa_rep_std / np.sqrt(n_measurements)
|
|
201
|
+
|
|
202
|
+
# Calculate 95% CI using t-distribution
|
|
203
|
+
df_actual = n_measurements - 1
|
|
204
|
+
t_critical = scipy_stats.t.ppf(0.975, df_actual)
|
|
205
|
+
aa_margin_of_error = t_critical * sem
|
|
206
|
+
|
|
207
|
+
aa_scores['SD_rep'] = aa_rep_std
|
|
208
|
+
aa_scores['CV_rep'] = (aa_rep_std / aa_rep_mean).round(3)
|
|
209
|
+
aa_scores['n_measurements'] = n_measurements.astype('Int64')
|
|
210
|
+
aa_scores['SEM'] = sem
|
|
211
|
+
aa_scores['CI_lower'] = aa_rep_mean - aa_margin_of_error
|
|
212
|
+
aa_scores['CI_upper'] = aa_rep_mean + aa_margin_of_error
|
|
213
|
+
|
|
214
|
+
return aa_scores
|
|
@@ -0,0 +1,188 @@
|
|
|
1
|
+
"""
|
|
2
|
+
Sequence annotation utilities for Sort-seq variant analysis.
|
|
3
|
+
|
|
4
|
+
This module provides functions for annotating variant DataFrames with sequence differences,
|
|
5
|
+
translations, and other derived sequence information.
|
|
6
|
+
|
|
7
|
+
Examples
|
|
8
|
+
--------
|
|
9
|
+
>>> from sortscore.analysis.annotation import annotate_scores_dataframe
|
|
10
|
+
>>> annotated_df = annotate_scores_dataframe(scores_df, experiment)
|
|
11
|
+
"""
|
|
12
|
+
import pandas as pd
|
|
13
|
+
from sortscore.utils.sequence_parsing import compare_to_reference, compare_codon_lists, translate_dna
|
|
14
|
+
|
|
15
|
+
NO_DIFF_MARKER = '='
|
|
16
|
+
|
|
17
|
+
|
|
18
|
+
# TODO: #37 redundant, see if we can remove
|
|
19
|
+
def annotate_scores_dataframe(
|
|
20
|
+
scores_df: pd.DataFrame,
|
|
21
|
+
wt_dna_seq: str,
|
|
22
|
+
mutagenesis_type: str = 'aa',
|
|
23
|
+
) -> pd.DataFrame:
|
|
24
|
+
"""
|
|
25
|
+
Add sequence annotation columns to a scores DataFrame.
|
|
26
|
+
|
|
27
|
+
Parameters
|
|
28
|
+
----------
|
|
29
|
+
scores_df : pd.DataFrame
|
|
30
|
+
DataFrame with variant sequences and scores.
|
|
31
|
+
wt_dna_seq : str
|
|
32
|
+
Wild-type DNA reference sequence.
|
|
33
|
+
mutagenesis_type : str, default 'aa'
|
|
34
|
+
Type of mutagenesis ('codon', 'snv', 'aa').
|
|
35
|
+
|
|
36
|
+
Returns
|
|
37
|
+
-------
|
|
38
|
+
annotated_df : pd.DataFrame
|
|
39
|
+
DataFrame with added annotation columns.
|
|
40
|
+
|
|
41
|
+
Examples
|
|
42
|
+
--------
|
|
43
|
+
>>> annotated_df = annotate_scores_dataframe(scores_df, wt_seq, 'dna')
|
|
44
|
+
"""
|
|
45
|
+
df = scores_df.copy()
|
|
46
|
+
|
|
47
|
+
# Check if aa_seq_diff already exists (from pre-annotated data)
|
|
48
|
+
has_pre_annotated_aa = 'aa_seq_diff' in df.columns
|
|
49
|
+
|
|
50
|
+
# Treat 'dna' as a DNA-sequence variant type (full-length DNA sequences)
|
|
51
|
+
if mutagenesis_type in {'codon', 'snv'}:
|
|
52
|
+
# Add codon differences
|
|
53
|
+
df['codon_diff'] = df['variant_seq'].apply(
|
|
54
|
+
lambda x: compare_codon_lists(wt_dna_seq, x)
|
|
55
|
+
)
|
|
56
|
+
df['codon_diff'] = df['codon_diff'].fillna('')
|
|
57
|
+
|
|
58
|
+
# Add DNA sequence differences
|
|
59
|
+
df['dna_seq_diff'] = df['variant_seq'].apply(
|
|
60
|
+
lambda x: compare_to_reference(wt_dna_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
61
|
+
)
|
|
62
|
+
|
|
63
|
+
# Add AA sequence annotations only if not pre-annotated
|
|
64
|
+
if not has_pre_annotated_aa:
|
|
65
|
+
wt_aa_seq = translate_dna(wt_dna_seq)
|
|
66
|
+
df['aa_seq'] = df['variant_seq'].apply(translate_dna)
|
|
67
|
+
df['aa_seq_diff'] = df['aa_seq'].apply(
|
|
68
|
+
lambda x: compare_to_reference(wt_aa_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
69
|
+
)
|
|
70
|
+
|
|
71
|
+
elif mutagenesis_type == 'aa':
|
|
72
|
+
# For AA variants, add sequence differences only if not pre-annotated
|
|
73
|
+
if not has_pre_annotated_aa:
|
|
74
|
+
wt_aa_seq = translate_dna(wt_dna_seq) if len(wt_dna_seq) % 3 == 0 else wt_dna_seq
|
|
75
|
+
df['aa_seq_diff'] = df['variant_seq'].apply(
|
|
76
|
+
lambda x: compare_to_reference(wt_aa_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
77
|
+
)
|
|
78
|
+
|
|
79
|
+
# Map stop codon representations to * for standard notation in aa_seq_diff column
|
|
80
|
+
if 'aa_seq_diff' in df.columns:
|
|
81
|
+
df['aa_seq_diff'] = df['aa_seq_diff'].str.replace('X', '*', regex=False)
|
|
82
|
+
df['aa_seq_diff'] = df['aa_seq_diff'].str.replace('Ter', '*', regex=False)
|
|
83
|
+
|
|
84
|
+
# Add functional annotations
|
|
85
|
+
df = add_variant_categories(df)
|
|
86
|
+
|
|
87
|
+
return df
|
|
88
|
+
|
|
89
|
+
# TODO: #37 isn't this redundant with similar functions
|
|
90
|
+
def add_sequence_differences(
|
|
91
|
+
df: pd.DataFrame,
|
|
92
|
+
wt_dna_seq: str,
|
|
93
|
+
mutagenesis_type: str = 'aa',
|
|
94
|
+
) -> pd.DataFrame:
|
|
95
|
+
"""
|
|
96
|
+
Add sequence difference columns to a DataFrame.
|
|
97
|
+
|
|
98
|
+
Parameters
|
|
99
|
+
----------
|
|
100
|
+
df : pd.DataFrame
|
|
101
|
+
DataFrame with variant sequences.
|
|
102
|
+
wt_dna_seq : str
|
|
103
|
+
Wild-type DNA sequence.
|
|
104
|
+
mutagenesis_type : str, default 'aa'
|
|
105
|
+
Type of mutagenesis ('codon', 'snv', 'aa').
|
|
106
|
+
|
|
107
|
+
Returns
|
|
108
|
+
-------
|
|
109
|
+
df : pd.DataFrame
|
|
110
|
+
DataFrame with sequence difference columns added.
|
|
111
|
+
"""
|
|
112
|
+
df = df.copy()
|
|
113
|
+
|
|
114
|
+
if mutagenesis_type in {'codon', 'snv'}:
|
|
115
|
+
# Add DNA sequence differences
|
|
116
|
+
df['dna_seq_diff'] = df['variant_seq'].apply(
|
|
117
|
+
lambda x: compare_to_reference(wt_dna_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
118
|
+
)
|
|
119
|
+
|
|
120
|
+
# Add AA sequence differences
|
|
121
|
+
wt_aa_seq = translate_dna(wt_dna_seq)
|
|
122
|
+
df['aa_seq'] = df['variant_seq'].apply(translate_dna)
|
|
123
|
+
df['aa_seq_diff'] = df['aa_seq'].apply(
|
|
124
|
+
lambda x: compare_to_reference(wt_aa_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
125
|
+
)
|
|
126
|
+
|
|
127
|
+
elif mutagenesis_type == 'aa':
|
|
128
|
+
# For AA variants, sequences are already amino acids
|
|
129
|
+
wt_aa_seq = translate_dna(wt_dna_seq) if len(wt_dna_seq) % 3 == 0 else wt_dna_seq
|
|
130
|
+
df['aa_seq_diff'] = df['variant_seq'].apply(
|
|
131
|
+
lambda x: compare_to_reference(wt_aa_seq, x, no_change_marker=NO_DIFF_MARKER)
|
|
132
|
+
)
|
|
133
|
+
|
|
134
|
+
return df
|
|
135
|
+
|
|
136
|
+
def classify_aa_variant(aa_diff, dna_diff=None):
|
|
137
|
+
if aa_diff == NO_DIFF_MARKER:
|
|
138
|
+
# Check if this is true WT (no DNA changes) or synonymous (DNA changes but same AA)
|
|
139
|
+
if dna_diff == NO_DIFF_MARKER:
|
|
140
|
+
return 'wt_dna'
|
|
141
|
+
else:
|
|
142
|
+
return 'synonymous'
|
|
143
|
+
elif '*' in aa_diff:
|
|
144
|
+
return 'nonsense'
|
|
145
|
+
else:
|
|
146
|
+
return 'missense_aa'
|
|
147
|
+
|
|
148
|
+
def classify_dna_variant(dna_diff, aa_diff):
|
|
149
|
+
if dna_diff == NO_DIFF_MARKER:
|
|
150
|
+
return 'wt_dna'
|
|
151
|
+
elif aa_diff == NO_DIFF_MARKER:
|
|
152
|
+
return 'synonymous'
|
|
153
|
+
else:
|
|
154
|
+
return 'missense_dna'
|
|
155
|
+
|
|
156
|
+
|
|
157
|
+
def add_variant_categories(df: pd.DataFrame) -> pd.DataFrame:
|
|
158
|
+
"""
|
|
159
|
+
Add variant category annotations based on existing sequence difference columns.
|
|
160
|
+
|
|
161
|
+
Parameters
|
|
162
|
+
----------
|
|
163
|
+
df : pd.DataFrame
|
|
164
|
+
DataFrame with sequence difference columns (aa_seq_diff, dna_seq_diff).
|
|
165
|
+
|
|
166
|
+
Returns
|
|
167
|
+
-------
|
|
168
|
+
df : pd.DataFrame
|
|
169
|
+
DataFrame with variant category columns added.
|
|
170
|
+
"""
|
|
171
|
+
df = df.copy()
|
|
172
|
+
|
|
173
|
+
# Classify DNA variants
|
|
174
|
+
if 'dna_seq_diff' in df.columns:
|
|
175
|
+
if 'aa_seq_diff' in df.columns:
|
|
176
|
+
df['annotate_dna'] = df.apply(lambda row: classify_dna_variant(row['dna_seq_diff'], row['aa_seq_diff']), axis=1)
|
|
177
|
+
else:
|
|
178
|
+
df['annotate_dna'] = df['dna_seq_diff'].apply(lambda x: 'missense_dna' if x else 'wt_dna')
|
|
179
|
+
|
|
180
|
+
|
|
181
|
+
# Classify variants based on AA changes
|
|
182
|
+
if 'aa_seq_diff' in df.columns:
|
|
183
|
+
if 'dna_seq_diff' in df.columns:
|
|
184
|
+
df['annotate_aa'] = df.apply(lambda row: classify_aa_variant(row['aa_seq_diff'], row['dna_seq_diff']), axis=1)
|
|
185
|
+
else:
|
|
186
|
+
df['annotate_aa'] = df['aa_seq_diff'].apply(classify_aa_variant)
|
|
187
|
+
|
|
188
|
+
return df
|