molscope 0.8.0__tar.gz → 0.8.2__tar.gz
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- molscope-0.8.2/CITATION.cff +15 -0
- molscope-0.8.2/MANIFEST.in +13 -0
- {molscope-0.8.0 → molscope-0.8.2}/PKG-INFO +220 -73
- {molscope-0.8.0 → molscope-0.8.2}/README.md +210 -71
- molscope-0.8.2/docs/api-reference.md +53 -0
- molscope-0.8.2/docs/assets/coarsegrain/1fqy-cg-mapping-comparison.png +0 -0
- molscope-0.8.2/docs/assets/coarsegrain/1fqy-martini-mapping.png +0 -0
- molscope-0.8.2/docs/assets/contactmaps/1aml-contact-frequency.png +0 -0
- molscope-0.8.2/docs/assets/contactmaps/1fqy-ca-distance-matrix.png +0 -0
- molscope-0.8.2/docs/assets/contactmaps/1fqy-residue-contact-map.png +0 -0
- molscope-0.8.2/docs/assets/geometry/1aml-rmsf-profile.png +0 -0
- molscope-0.8.2/docs/assets/geometry/1fqy-principal-axes.png +0 -0
- molscope-0.8.2/docs/assets/graphs/1fqy-residue-contact-graph.png +0 -0
- molscope-0.8.2/docs/assets/readme/aquaporin-structure-v2.png +0 -0
- molscope-0.8.2/docs/assets/readme/coarse-grained-beads-v2.png +0 -0
- molscope-0.8.2/docs/assets/readme/residue-contact-map.png +0 -0
- molscope-0.8.2/docs/assets/readme/secondary-structure.png +0 -0
- molscope-0.8.2/docs/benchmarks.md +55 -0
- molscope-0.8.2/docs/contributing.md +32 -0
- molscope-0.8.2/docs/examples/analyze-contacts.md +42 -0
- molscope-0.8.2/docs/examples/binding-site.md +36 -0
- molscope-0.8.2/docs/examples/build-molecular-graph.md +25 -0
- molscope-0.8.2/docs/examples/coarse-grain-protein.md +61 -0
- molscope-0.8.2/docs/examples/compare-nmr-models.md +15 -0
- molscope-0.8.2/docs/examples/export-pyg.md +25 -0
- molscope-0.8.2/docs/examples/geometry-tour.md +37 -0
- molscope-0.8.2/docs/examples/index.md +32 -0
- molscope-0.8.2/docs/examples/pdb-to-graph-cg.md +78 -0
- molscope-0.8.2/docs/examples/pdb-to-pyg-ml.md +114 -0
- molscope-0.8.2/docs/examples/protein-analysis-from-scratch.md +56 -0
- molscope-0.8.2/docs/examples/residue-contact-graphs.md +40 -0
- molscope-0.8.2/docs/index.md +56 -0
- molscope-0.8.2/docs/installation.md +53 -0
- molscope-0.8.2/docs/limitations.md +191 -0
- molscope-0.8.2/docs/quickstart.md +56 -0
- molscope-0.8.2/docs/roadmap.md +56 -0
- molscope-0.8.2/docs/tutorials/index.md +16 -0
- molscope-0.8.2/docs/tutorials/pdb-to-coarse-grained-beads.md +207 -0
- molscope-0.8.2/docs/tutorials/pdb-to-descriptors.md +138 -0
- molscope-0.8.2/docs/tutorials/pdb-to-graph-gnn.md +180 -0
- molscope-0.8.2/docs/user-guide/chemical-perception.md +35 -0
- molscope-0.8.2/docs/user-guide/coarse-graining.md +210 -0
- molscope-0.8.2/docs/user-guide/contact-maps.md +152 -0
- molscope-0.8.2/docs/user-guide/coordinate-formats.md +122 -0
- molscope-0.8.2/docs/user-guide/descriptors.md +85 -0
- molscope-0.8.2/docs/user-guide/ensembles.md +33 -0
- molscope-0.8.2/docs/user-guide/geometry.md +123 -0
- molscope-0.8.2/docs/user-guide/molecular-graphs.md +121 -0
- molscope-0.8.2/docs/user-guide/plotting.md +33 -0
- molscope-0.8.2/docs/user-guide/protein-analysis.md +154 -0
- molscope-0.8.2/docs/user-guide/reading-files.md +63 -0
- molscope-0.8.2/docs/user-guide/selections.md +33 -0
- molscope-0.8.2/docs/validation.md +91 -0
- molscope-0.8.2/examples/binding_site.py +44 -0
- molscope-0.8.2/examples/coarse_graining.py +82 -0
- molscope-0.8.2/examples/data/1aml.pdb +12391 -0
- molscope-0.8.2/examples/data/1fqy.pdb +2083 -0
- molscope-0.8.2/examples/data/3ptb.pdb +2264 -0
- molscope-0.8.2/examples/data/helix_201.xyz +203 -0
- molscope-0.8.2/examples/geometry.py +59 -0
- molscope-0.8.2/examples/graph_to_gnn.py +64 -0
- molscope-0.8.2/examples/legacy_utils.py +56 -0
- molscope-0.8.2/examples/pdb_to_pyg_ml.py +136 -0
- molscope-0.8.2/examples/protein_analysis.py +92 -0
- molscope-0.8.2/examples/residue_contact_graph.py +121 -0
- molscope-0.8.2/examples/tour.py +119 -0
- molscope-0.8.2/mkdocs.yml +63 -0
- molscope-0.8.2/molscope/__init__.py +158 -0
- molscope-0.8.2/molscope/cli.py +330 -0
- molscope-0.8.2/molscope/coarsegrain.py +917 -0
- molscope-0.8.2/molscope/contactmap.py +182 -0
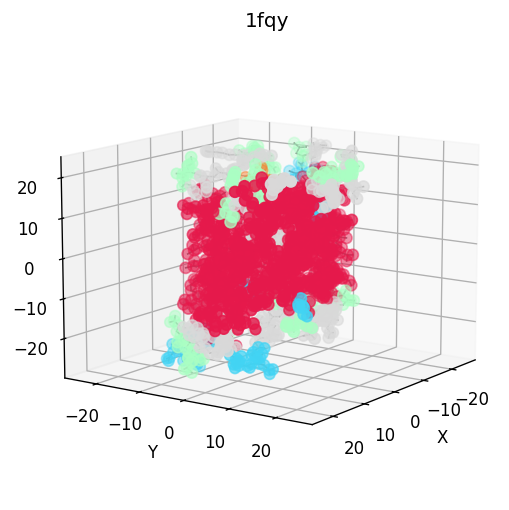
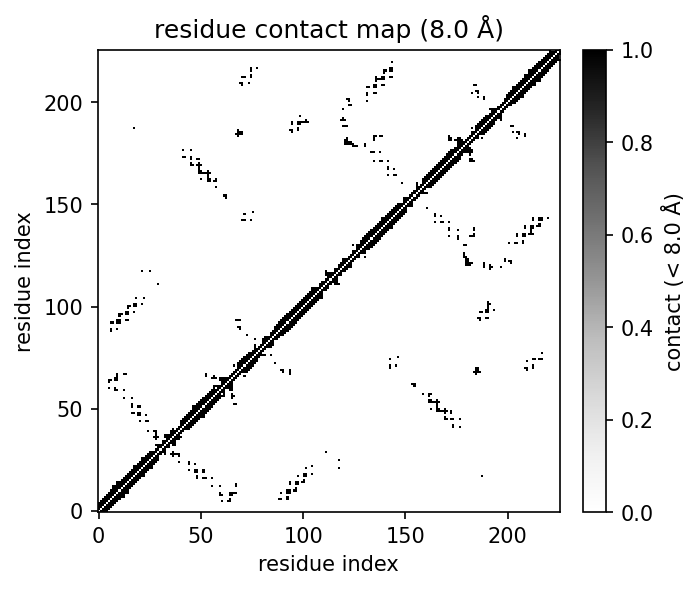
- molscope-0.8.2/molscope/contacts.py +292 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/descriptors.py +4 -3
- molscope-0.8.2/molscope/distance.py +438 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/dssp.py +135 -12
- {molscope-0.8.0 → molscope-0.8.2}/molscope/ensemble.py +15 -3
- molscope-0.8.2/molscope/graph.py +1011 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/io.py +259 -73
- {molscope-0.8.0 → molscope-0.8.2}/molscope/molecule.py +447 -108
- molscope-0.8.2/molscope/plotting.py +365 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope.egg-info/PKG-INFO +220 -73
- molscope-0.8.2/molscope.egg-info/SOURCES.txt +135 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope.egg-info/requires.txt +8 -0
- molscope-0.8.2/notebooks/molscope_tour.ipynb +448 -0
- molscope-0.8.2/notebooks/pdb_to_gnn.ipynb +276 -0
- molscope-0.8.2/notebooks/protein_analysis_from_scratch.ipynb +302 -0
- {molscope-0.8.0 → molscope-0.8.2}/pyproject.toml +35 -5
- molscope-0.8.2/requirements.txt +2 -0
- molscope-0.8.2/scripts/benchmark_core.py +129 -0
- molscope-0.8.2/scripts/build_gnn_notebook.py +242 -0
- molscope-0.8.2/scripts/build_protein_analysis_notebook.py +250 -0
- molscope-0.8.2/scripts/build_user_guide_pdf.py +73 -0
- molscope-0.8.2/scripts/render_coarsegrain_images.py +59 -0
- molscope-0.8.2/scripts/render_contact_analysis_images.py +48 -0
- molscope-0.8.2/scripts/render_geometry_images.py +81 -0
- molscope-0.8.2/tests/fixtures/bad_coord.pdb +3 -0
- molscope-0.8.2/tests/fixtures/bad_coord.xyz +4 -0
- molscope-0.8.2/tests/fixtures/bad_counts.sdf +6 -0
- molscope-0.8.2/tests/fixtures/missing_coord_col.cif +10 -0
- molscope-0.8.2/tests/fixtures/no_atom_site.cif +10 -0
- molscope-0.8.2/tests/fixtures/no_atoms.pdb +3 -0
- molscope-0.8.2/tests/fixtures/short_atom.pdb +3 -0
- molscope-0.8.2/tests/fixtures/truncated.sdf +6 -0
- molscope-0.8.2/tests/fixtures/truncated.xyz +4 -0
- molscope-0.8.2/tests/fixtures/v3000.sdf +15 -0
- molscope-0.8.2/tests/fixtures/water.sdf +11 -0
- molscope-0.8.2/tests/test_cg_mapping.py +215 -0
- molscope-0.8.2/tests/test_cif_validation.py +153 -0
- molscope-0.8.2/tests/test_cli.py +48 -0
- molscope-0.8.2/tests/test_cli_batch.py +54 -0
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_clustering.py +1 -1
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_coarsegrain.py +82 -4
- molscope-0.8.2/tests/test_contactmap.py +163 -0
- molscope-0.8.2/tests/test_contacts.py +122 -0
- molscope-0.8.2/tests/test_dssp.py +120 -0
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_extras.py +1 -0
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_features.py +28 -1
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_graph.py +157 -3
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_io.py +118 -5
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_molecule.py +11 -12
- molscope-0.8.2/tests/test_protein_workflows.py +58 -0
- molscope-0.8.2/tests/validation/test_bonds_ref.py +56 -0
- molscope-0.8.2/tests/validation/test_chem_ref.py +95 -0
- molscope-0.8.2/tests/validation/test_dssp_ref.py +125 -0
- molscope-0.8.2/tests/validation/test_geometry_ref.py +135 -0
- molscope-0.8.2/tests/validation/test_invariants.py +109 -0
- molscope-0.8.0/molscope/__init__.py +0 -108
- molscope-0.8.0/molscope/cli.py +0 -74
- molscope-0.8.0/molscope/coarsegrain.py +0 -411
- molscope-0.8.0/molscope/contactmap.py +0 -118
- molscope-0.8.0/molscope/graph.py +0 -305
- molscope-0.8.0/molscope/plotting.py +0 -196
- molscope-0.8.0/molscope.egg-info/SOURCES.txt +0 -36
- molscope-0.8.0/tests/test_cif_validation.py +0 -69
- molscope-0.8.0/tests/test_contactmap.py +0 -87
- molscope-0.8.0/tests/test_dssp.py +0 -67
- {molscope-0.8.0 → molscope-0.8.2}/LICENSE +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/__main__.py +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/chem.py +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/cif.py +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope/elements.py +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope.egg-info/dependency_links.txt +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope.egg-info/entry_points.txt +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/molscope.egg-info/top_level.txt +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/setup.cfg +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_chem.py +0 -0
- {molscope-0.8.0 → molscope-0.8.2}/tests/test_descriptors.py +0 -0
|
@@ -0,0 +1,15 @@
|
|
|
1
|
+
cff-version: 1.2.0
|
|
2
|
+
message: "If you use MolScope in teaching, research, or software demos, please cite it."
|
|
3
|
+
type: software
|
|
4
|
+
title: "MolScope: lightweight molecular structure analysis, visualisation, graph export, and coarse-graining in Python"
|
|
5
|
+
authors:
|
|
6
|
+
- family-names: Shrestha
|
|
7
|
+
given-names: Roshan
|
|
8
|
+
version: 0.8.2
|
|
9
|
+
date-released: "2026-05-28"
|
|
10
|
+
license: MIT
|
|
11
|
+
repository-code: "https://github.com/roshan2004/molscope"
|
|
12
|
+
url: "https://github.com/roshan2004/molscope"
|
|
13
|
+
abstract: >
|
|
14
|
+
MolScope is a lightweight Python toolkit for molecular structure analysis,
|
|
15
|
+
visualisation, descriptors, graph export, and coarse-graining workflows.
|
|
@@ -0,0 +1,13 @@
|
|
|
1
|
+
include CITATION.cff
|
|
2
|
+
include LICENSE
|
|
3
|
+
include README.md
|
|
4
|
+
include mkdocs.yml
|
|
5
|
+
include requirements.txt
|
|
6
|
+
recursive-include docs *.md *.png
|
|
7
|
+
recursive-include examples *.py
|
|
8
|
+
recursive-include examples/data *.pdb *.xyz
|
|
9
|
+
recursive-include notebooks *.ipynb
|
|
10
|
+
recursive-include scripts *.py
|
|
11
|
+
recursive-include tests *.py
|
|
12
|
+
recursive-include tests/fixtures *.xyz *.pdb *.cif *.sdf
|
|
13
|
+
prune notebooks/.ipynb_checkpoints
|
|
@@ -1,11 +1,13 @@
|
|
|
1
1
|
Metadata-Version: 2.4
|
|
2
2
|
Name: molscope
|
|
3
|
-
Version: 0.8.
|
|
4
|
-
Summary: Lightweight molecular
|
|
3
|
+
Version: 0.8.2
|
|
4
|
+
Summary: Lightweight molecular coordinate workflows for descriptors, graph ML, and coarse-grained beads.
|
|
5
5
|
Author-email: Roshan Shrestha <roshanpra@gmail.com>
|
|
6
6
|
License-Expression: MIT
|
|
7
7
|
Project-URL: Homepage, https://github.com/roshan2004/molscope
|
|
8
8
|
Project-URL: Repository, https://github.com/roshan2004/molscope
|
|
9
|
+
Project-URL: Documentation, https://molscope.readthedocs.io/
|
|
10
|
+
Project-URL: Issues, https://github.com/roshan2004/molscope/issues
|
|
9
11
|
Keywords: chemistry,molecular-structure,pdb,xyz,mmcif,coarse-graining,molecular-graphs,machine-learning,visualization
|
|
10
12
|
Classifier: Programming Language :: Python :: 3
|
|
11
13
|
Classifier: Topic :: Scientific/Engineering :: Chemistry
|
|
@@ -17,6 +19,7 @@ Requires-Dist: numpy>=1.21
|
|
|
17
19
|
Requires-Dist: matplotlib>=3.5
|
|
18
20
|
Provides-Extra: test
|
|
19
21
|
Requires-Dist: pytest>=7; extra == "test"
|
|
22
|
+
Requires-Dist: pytest-cov>=4; extra == "test"
|
|
20
23
|
Provides-Extra: fast
|
|
21
24
|
Requires-Dist: scipy>=1.7; extra == "fast"
|
|
22
25
|
Provides-Extra: viz
|
|
@@ -27,6 +30,11 @@ Provides-Extra: chem
|
|
|
27
30
|
Requires-Dist: rdkit>=2023.9; extra == "chem"
|
|
28
31
|
Provides-Extra: cif
|
|
29
32
|
Requires-Dist: gemmi>=0.7; extra == "cif"
|
|
33
|
+
Provides-Extra: gpu
|
|
34
|
+
Requires-Dist: torch>=2.0; extra == "gpu"
|
|
35
|
+
Provides-Extra: validation
|
|
36
|
+
Requires-Dist: mdanalysis>=2.7; extra == "validation"
|
|
37
|
+
Requires-Dist: rdkit>=2023.9; extra == "validation"
|
|
30
38
|
Provides-Extra: pyg
|
|
31
39
|
Requires-Dist: torch>=2.0; extra == "pyg"
|
|
32
40
|
Requires-Dist: torch-geometric>=2.3; extra == "pyg"
|
|
@@ -43,58 +51,105 @@ Dynamic: license-file
|
|
|
43
51
|
# MolScope
|
|
44
52
|
|
|
45
53
|
[](https://github.com/roshan2004/molscope/actions/workflows/ci.yml)
|
|
54
|
+
[](https://codecov.io/gh/roshan2004/molscope)
|
|
55
|
+
[](https://molscope.readthedocs.io/en/latest/)
|
|
56
|
+
[](https://pypi.org/project/molscope/)
|
|
46
57
|
[](pyproject.toml)
|
|
47
58
|
[](LICENSE)
|
|
48
59
|
[](https://github.com/astral-sh/ruff)
|
|
49
60
|
|
|
50
|
-
Lightweight molecular structure analysis,
|
|
51
|
-
|
|
52
|
-
|
|
53
|
-
|
|
54
|
-
|
|
55
|
-
`
|
|
61
|
+
Lightweight molecular structure analysis, graph export, and coarse-graining in
|
|
62
|
+
Python. MolScope is built around three polished workflows: turn coordinate
|
|
63
|
+
files into descriptor tables, graph-ML inputs, or coarse-grained bead
|
|
64
|
+
representations without pulling in a full simulation stack.
|
|
65
|
+
|
|
66
|
+
Read `.xyz`, `.pdb`, `.cif` and `.sdf` files, select and analyse atoms, and
|
|
67
|
+
visualise structures in 3D. Optional extras add Gemmi validation, RDKit chemical
|
|
68
|
+
features, PyTorch Geometric, DGL, and other backend integrations only when a
|
|
69
|
+
workflow needs them.
|
|
56
70
|
|
|
57
71
|
| 3D structure (element) | Secondary structure (DSSP) | Residue contact map | Coarse-grained beads |
|
|
58
72
|
| --- | --- | --- | --- |
|
|
59
73
|
|  |  |  | 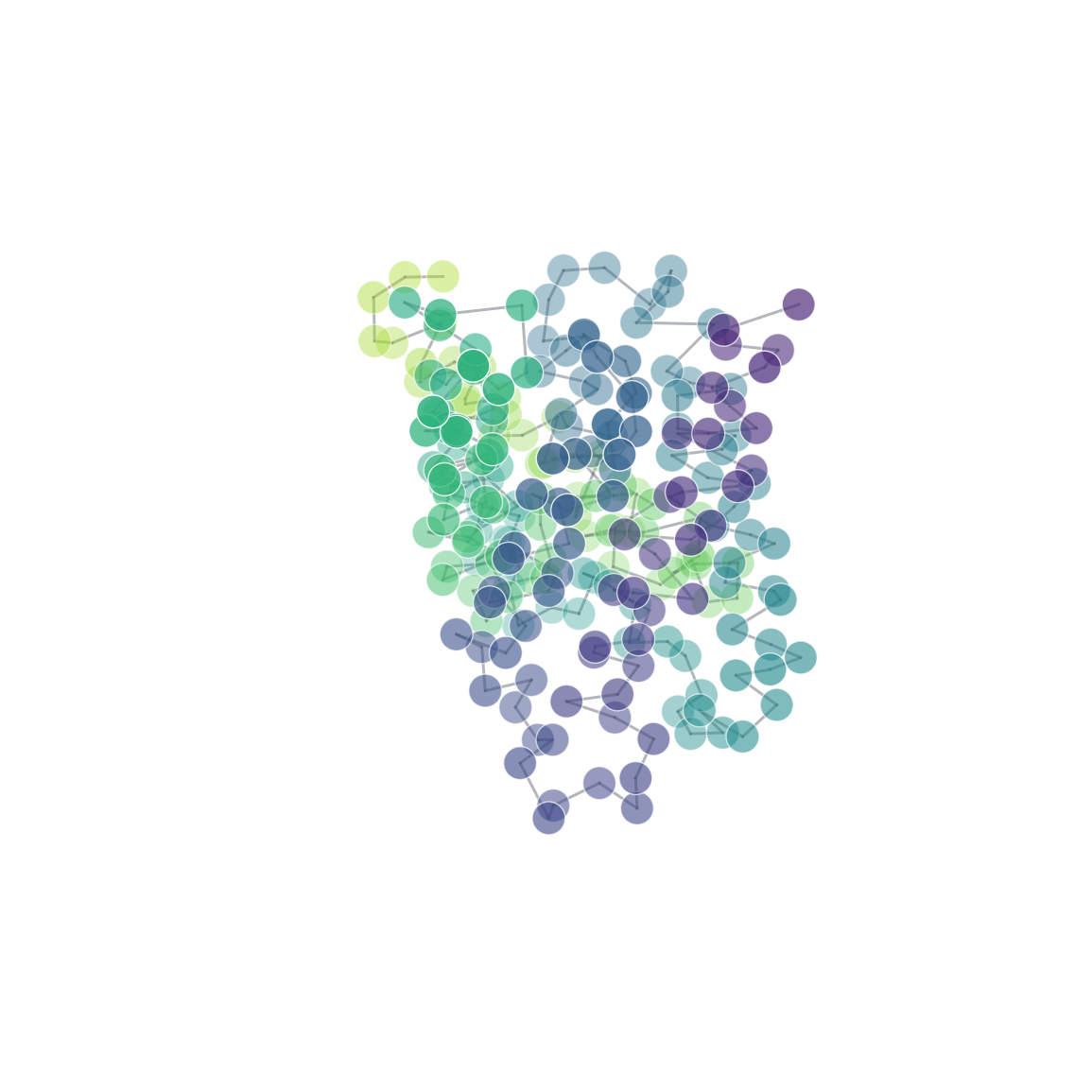 |
|
|
60
74
|
|
|
61
|
-
##
|
|
62
|
-
|
|
63
|
-
- **Read and write** XYZ, PDB, mmCIF and SDF (gzip-aware), preserve SDF/PDB
|
|
64
|
-
explicit bonds and SDF formal charges where present, fetch structures by id
|
|
65
|
-
from RCSB, and load multi-model NMR ensembles.
|
|
66
|
-
- **Validate mmCIF** syntax, atom-site coordinate columns, and supplied
|
|
67
|
-
dictionary files with optional Gemmi support.
|
|
68
|
-
- **Select and measure** by chain, element or residue; compute distances,
|
|
69
|
-
angles, dihedrals and Kabsch-aligned RMSD.
|
|
70
|
-
- **Analyse** centroids, radius of gyration, the inertia tensor,
|
|
71
|
-
explicit/inferred bonds, and contacts.
|
|
72
|
-
- **Contact maps** at atom or residue level, with heatmap plots.
|
|
73
|
-
- **Secondary structure** via a self-contained, dependency-free DSSP, with
|
|
74
|
-
`plot(color_by="ss")`.
|
|
75
|
-
- **Ensembles**: pairwise RMSD, RMSF, averaging, and conformer clustering.
|
|
76
|
-
- **Export for ML**: flat structural descriptors and molecular graphs for
|
|
77
|
-
NetworkX, PyTorch Geometric and DGL.
|
|
78
|
-
- **Chemical perception and descriptors**: optional RDKit-backed formal charge,
|
|
79
|
-
valence, aromaticity and scalar descriptor features with
|
|
80
|
-
`pip install "molscope[chem]"`.
|
|
81
|
-
- **Coarse-grain** onto residue, Martini-style or custom bead mappings.
|
|
82
|
-
- **Visualise** with 3D matplotlib plots, an interactive py3Dmol viewer, spin
|
|
83
|
-
GIFs, and a command-line interface.
|
|
75
|
+
## Core workflows
|
|
84
76
|
|
|
85
|
-
|
|
77
|
+
| Workflow | Start here | Output |
|
|
78
|
+
| --- | --- | --- |
|
|
79
|
+
| **PDB to descriptors** | [`docs/tutorials/pdb-to-descriptors.md`](docs/tutorials/pdb-to-descriptors.md) | Fixed-width structural and optional RDKit-backed feature tables for screening, QC, and classical ML. |
|
|
80
|
+
| **PDB to graph/GNN** | [`docs/tutorials/pdb-to-graph-gnn.md`](docs/tutorials/pdb-to-graph-gnn.md) | Atom/bond, residue-contact, and PyTorch Geometric-ready graph data for message-passing experiments. |
|
|
81
|
+
| **PDB to coarse-grained beads** | [`docs/tutorials/pdb-to-coarse-grained-beads.md`](docs/tutorials/pdb-to-coarse-grained-beads.md) | Residue, simplified Martini-style, custom, and virtual-site bead models for inspection and graph prototyping. |
|
|
82
|
+
|
|
83
|
+
## Who is this for?
|
|
84
|
+
|
|
85
|
+
- Students learning molecular-coordinate formats, structure analysis, and basic
|
|
86
|
+
visualisation from readable Python code.
|
|
87
|
+
- Molecular modellers who want quick static-structure checks, selections,
|
|
88
|
+
contact maps, and lightweight coarse-grained mapping prototypes.
|
|
89
|
+
- ML-for-molecules learners who need deterministic descriptors and graph exports
|
|
90
|
+
before moving to larger chemistry or simulation frameworks.
|
|
91
|
+
|
|
92
|
+
## Supporting capabilities
|
|
93
|
+
|
|
94
|
+
MolScope keeps the broad feature surface organized around those workflows:
|
|
95
|
+
|
|
96
|
+
- **Input layer**: read and write XYZ, PDB, mmCIF and SDF, preserve explicit
|
|
97
|
+
bonds and charges where present, fetch structures from RCSB, and validate
|
|
98
|
+
mmCIF files with optional Gemmi support.
|
|
99
|
+
- **Structure analysis**: select atoms by metadata, compute geometry, RMSD,
|
|
100
|
+
contacts, contact maps, interfaces, binding sites, and ensemble summaries.
|
|
101
|
+
- **Annotations and descriptors**: run dependency-free secondary-structure
|
|
102
|
+
assignment, native structural descriptors, and optional RDKit-backed
|
|
103
|
+
chemistry descriptors.
|
|
104
|
+
- **Representation outputs**: export descriptor tables, atom/bond graphs,
|
|
105
|
+
residue-contact graphs, and coarse-grained bead graphs to common ML backends.
|
|
106
|
+
- **Inspection and automation**: visualize with Matplotlib or py3Dmol and use
|
|
107
|
+
the CLI for view, analyze, and export workflows.
|
|
108
|
+
|
|
109
|
+
## Architecture
|
|
110
|
+
|
|
111
|
+
```mermaid
|
|
112
|
+
graph LR
|
|
113
|
+
A[XYZ, PDB, mmCIF, SDF files] --> B[read, fetch, validate]
|
|
114
|
+
B --> C[Molecule object]
|
|
115
|
+
C --> D[Geometry, contacts, DSSP, descriptors]
|
|
116
|
+
C --> E[Molecular graphs for NetworkX, PyG, DGL]
|
|
117
|
+
C --> F[Residue, simplified Martini-style, custom CG mappings]
|
|
118
|
+
C --> G[Matplotlib, py3Dmol, CLI visualisation]
|
|
119
|
+
```
|
|
86
120
|
|
|
87
|
-
|
|
88
|
-
cheminformatics frameworks. It is a lightweight **educational and prototyping**
|
|
89
|
-
toolkit for reading common molecular structure files, performing simple
|
|
90
|
-
structural analysis, exporting graph representations for ML workflows, and
|
|
91
|
-
experimenting with coarse-grained mappings. Its core depends only on NumPy and
|
|
92
|
-
Matplotlib, and the API is Python-first and scriptable.
|
|
121
|
+
## Why MolScope?
|
|
93
122
|
|
|
94
|
-
|
|
95
|
-
|
|
96
|
-
|
|
97
|
-
|
|
123
|
+
MolScope takes you from a structure file to a descriptor table, a
|
|
124
|
+
machine-learning graph, or a coarse-grained model with the smallest install that
|
|
125
|
+
gets the job done. The core depends only on NumPy and Matplotlib, so
|
|
126
|
+
`pip install molscope` stays light. Everything heavier (RDKit, PyTorch, PyTorch
|
|
127
|
+
Geometric, DGL, MDAnalysis, Gemmi) is an opt-in [extra](#install) you add only
|
|
128
|
+
when one of those workflows needs it.
|
|
129
|
+
|
|
130
|
+
That makes it an on-ramp rather than a framework:
|
|
131
|
+
|
|
132
|
+
- **Structure to ML graph in a few lines.** `mol.to_graph()` runs with zero extra
|
|
133
|
+
dependencies; `mol.to_pyg_data()`, `mol.to_networkx()` and `mol.to_dgl_graph()`
|
|
134
|
+
hand off to the ecosystem when you want it. This is the path MolScope is built
|
|
135
|
+
to make easy.
|
|
136
|
+
- **Descriptor tables without a framework.** Native structural descriptors and
|
|
137
|
+
optional RDKit-backed chemistry descriptors share the same `Molecule` and
|
|
138
|
+
batch-featurization APIs.
|
|
139
|
+
- **CG as a representation tool.** Residue, simplified Martini-style, custom
|
|
140
|
+
bead, and virtual-site mappings are inspectable molecules and bead graphs,
|
|
141
|
+
not hidden simulation setup state.
|
|
142
|
+
- **Light enough to teach and prototype with.** A readable, Python-first API over
|
|
143
|
+
static structures, with no trajectory engine or build step to wrestle.
|
|
144
|
+
- **Honest about its numbers.** Bonds, geometry and DSSP are cross-checked against
|
|
145
|
+
reference tools (MDAnalysis, `mkdssp`) in CI, so you know where the results come
|
|
146
|
+
from.
|
|
147
|
+
|
|
148
|
+
MolScope is **not** a replacement for full molecular-simulation or
|
|
149
|
+
cheminformatics frameworks. In particular, the coarse-graining tools are for
|
|
150
|
+
**educational CG mapping and bead-graph prototyping**: useful for exploring
|
|
151
|
+
mappings before moving to a production Martini workflow, not a validated Martini
|
|
152
|
+
force-field generator.
|
|
98
153
|
|
|
99
154
|
| Tool | Main focus | How MolScope differs |
|
|
100
155
|
| --- | --- | --- |
|
|
@@ -106,8 +161,8 @@ generator.
|
|
|
106
161
|
| nglview | Notebook structure viewer | MolScope also does analysis, descriptors, graphs and CG, not just viewing |
|
|
107
162
|
|
|
108
163
|
Reach for those tools when you need their depth and validation. Reach for
|
|
109
|
-
MolScope when you want
|
|
110
|
-
prototype
|
|
164
|
+
MolScope when you want the shortest path from a structure file to analysis, an
|
|
165
|
+
ML graph, or a CG prototype.
|
|
111
166
|
|
|
112
167
|
## Install
|
|
113
168
|
|
|
@@ -115,7 +170,7 @@ With [uv](https://docs.astral.sh/uv/) (recommended):
|
|
|
115
170
|
|
|
116
171
|
```bash
|
|
117
172
|
uv sync # creates .venv, installs deps + dev tools from the lockfile
|
|
118
|
-
uv run molscope 1fqy.pdb # run the CLI
|
|
173
|
+
uv run molscope examples/data/1fqy.pdb # run the CLI
|
|
119
174
|
uv run pytest # run the tests
|
|
120
175
|
```
|
|
121
176
|
|
|
@@ -125,6 +180,9 @@ uv run pytest # run the tests
|
|
|
125
180
|
With plain pip:
|
|
126
181
|
|
|
127
182
|
```bash
|
|
183
|
+
pip install molscope
|
|
184
|
+
|
|
185
|
+
# for local development from this checkout:
|
|
128
186
|
python -m venv .venv && source .venv/bin/activate
|
|
129
187
|
pip install -e ".[test]" # or: pip install -r requirements.txt
|
|
130
188
|
```
|
|
@@ -142,25 +200,49 @@ python scripts/build_user_guide_pdf.py
|
|
|
142
200
|
Docs source lives in `docs/`; the site configuration is `mkdocs.yml`. The PDF
|
|
143
201
|
builder requires Pandoc and a LaTeX engine such as `xelatex`.
|
|
144
202
|
|
|
203
|
+
Scientific validation is documented in
|
|
204
|
+
[`docs/validation.md`](docs/validation.md): reference tools, assumptions,
|
|
205
|
+
failure modes, and tolerances for MDAnalysis, RDKit, `mkdssp`, and invariant
|
|
206
|
+
checks.
|
|
207
|
+
|
|
145
208
|
## Quickstart
|
|
146
209
|
|
|
147
210
|
A runnable end-to-end tour over the bundled sample structures lives in
|
|
148
|
-
[`
|
|
211
|
+
[`examples/tour.py`](examples/tour.py):
|
|
149
212
|
|
|
150
213
|
```bash
|
|
151
|
-
uv run python
|
|
152
|
-
MPLBACKEND=Agg uv run python
|
|
214
|
+
uv run python examples/tour.py # opens 3D plot windows
|
|
215
|
+
MPLBACKEND=Agg uv run python examples/tour.py # headless: saves PNGs instead
|
|
153
216
|
```
|
|
154
217
|
|
|
155
218
|
It reads an `.xyz` and a `.pdb`, prints derived properties, compares the NMR
|
|
156
219
|
models of `1aml`, writes a transformed structure back out, and renders a plot.
|
|
157
220
|
|
|
221
|
+
For polished, focused workflows, start with the tutorials:
|
|
222
|
+
[`PDB to descriptors`](docs/tutorials/pdb-to-descriptors.md),
|
|
223
|
+
[`PDB to graph/GNN`](docs/tutorials/pdb-to-graph-gnn.md), and
|
|
224
|
+
[`PDB to coarse-grained beads`](docs/tutorials/pdb-to-coarse-grained-beads.md).
|
|
225
|
+
For coarse-graining as a runnable visual workflow, run
|
|
226
|
+
[`examples/coarse_graining.py`](examples/coarse_graining.py): residue
|
|
227
|
+
center-of-mass beads, residue centroids, and a simplified backbone/sidechain
|
|
228
|
+
mapping with a visual atomistic-to-CG comparison.
|
|
229
|
+
|
|
230
|
+
For protein-coordinate analysis from scratch, see
|
|
231
|
+
[`docs/examples/protein-analysis-from-scratch.md`](docs/examples/protein-analysis-from-scratch.md)
|
|
232
|
+
and [`notebooks/protein_analysis_from_scratch.ipynb`](notebooks/protein_analysis_from_scratch.ipynb):
|
|
233
|
+
backbone atoms, residues, chains, alpha carbons, contact maps, NMR ensemble
|
|
234
|
+
contacts, ligands, waters, binding sites, and simplified DSSP.
|
|
235
|
+
|
|
236
|
+
For a runnable, narrated version of that ML walkthrough, open the notebook
|
|
237
|
+
[`notebooks/pdb_to_gnn.ipynb`](notebooks/pdb_to_gnn.ipynb): structure file to a
|
|
238
|
+
trained GNN, end to end (needs `pip install 'molscope[pyg]'`).
|
|
239
|
+
|
|
158
240
|
## Library
|
|
159
241
|
|
|
160
242
|
```python
|
|
161
243
|
import molscope as ms
|
|
162
244
|
|
|
163
|
-
mol = ms.read("1fqy.pdb")
|
|
245
|
+
mol = ms.read("examples/data/1fqy.pdb") # parser chosen from the extension
|
|
164
246
|
mol = ms.fetch("1fqy") # ...or download straight from RCSB by id
|
|
165
247
|
print(mol.summary()) # atoms, formula, chains, bounding box
|
|
166
248
|
|
|
@@ -193,6 +275,7 @@ mol[mask_or_indices] # subset by numpy mask / index array
|
|
|
193
275
|
mol.centroid, mol.center_of_mass # geometric / mass-weighted centre
|
|
194
276
|
mol.radius_of_gyration # compactness (angstrom)
|
|
195
277
|
mol.dimensions, mol.formula # bounding box, Hill-order formula
|
|
278
|
+
mol.distance_matrix() # dense pairwise matrix (NumPy, Torch, CuPy)
|
|
196
279
|
mol.bonds() # inferred bond index pairs (KD-tree if scipy)
|
|
197
280
|
mol.contacts(cutoff=5.0) # atom pairs within a distance
|
|
198
281
|
mol.contact_count(cutoff=5.0) # count pairs without returning them
|
|
@@ -246,15 +329,17 @@ mol.plot_contact_map(cutoff=8.0) # heatmap
|
|
|
246
329
|
mol.contact_map(level="atom") # atom-level map
|
|
247
330
|
mol.contact_map(level="residue", method="min") # closest inter-residue atom
|
|
248
331
|
mol.contact_map(level="residue", method="com") # residue centre of mass
|
|
332
|
+
mol.contact_map(level="residue", backend="torch", device="cuda") # optional GPU
|
|
249
333
|
```
|
|
250
334
|
|
|
251
335
|
### Secondary structure (DSSP)
|
|
252
336
|
|
|
253
337
|
Assign protein secondary structure from backbone hydrogen-bond patterns with a
|
|
254
|
-
self-contained, pure-NumPy DSSP (no
|
|
338
|
+
self-contained, pure-NumPy, **simplified DSSP-style** implementation (no
|
|
339
|
+
external `mkdssp` binary needed):
|
|
255
340
|
|
|
256
341
|
```python
|
|
257
|
-
mol = ms.read("1fqy.pdb")
|
|
342
|
+
mol = ms.read("examples/data/1fqy.pdb")
|
|
258
343
|
ss = mol.secondary_structure() # SecondaryStructure, one code per residue
|
|
259
344
|
|
|
260
345
|
ss.string # e.g. '--HHHHHHHH--SS--EEEE--'
|
|
@@ -264,19 +349,23 @@ ss.summary() # helix/strand/coil counts and fractions
|
|
|
264
349
|
mol.plot(color_by="ss") # colour the 3D view by secondary structure
|
|
265
350
|
```
|
|
266
351
|
|
|
267
|
-
Codes follow DSSP: `H`/`G`/`I` helices, `E`/`B` strands, `T` turn, `S`
|
|
268
|
-
`-` coil. This is a simplified **educational** implementation
|
|
269
|
-
|
|
270
|
-
|
|
271
|
-
|
|
272
|
-
|
|
352
|
+
Codes follow DSSP notation: `H`/`G`/`I` helices, `E`/`B` strands, `T` turn, `S`
|
|
353
|
+
bend, `-` coil. This is a simplified **educational** implementation of the
|
|
354
|
+
Kabsch-Sander hydrogen-bond model: not bit-identical to the reference `mkdssp`
|
|
355
|
+
on every edge case, but validated against it. A CI cross-check
|
|
356
|
+
(`tests/validation`) puts it at **~99% per-residue 3-state agreement**
|
|
357
|
+
(helix/strand/coil) with `mkdssp` 4.2.2 on the bundled aquaporin (`1fqy`);
|
|
358
|
+
strand-rich folds, where reference DSSP is hardest to match, will agree less
|
|
359
|
+
closely. It needs backbone N/CA/C/O atoms, so use PDB/mmCIF input (not a bare
|
|
360
|
+
`.xyz`). The secondary-structure render in the showcase above (helices red,
|
|
361
|
+
turns cyan, coil grey) is produced this way.
|
|
273
362
|
|
|
274
363
|
### NMR ensembles
|
|
275
364
|
|
|
276
365
|
```python
|
|
277
366
|
from molscope import ensemble
|
|
278
367
|
|
|
279
|
-
models = ms.read_pdb_models("1aml.pdb") # all 20 models
|
|
368
|
+
models = ms.read_pdb_models("examples/data/1aml.pdb") # all 20 models
|
|
280
369
|
ensemble.rmsd_matrix(models) # pairwise RMSD matrix
|
|
281
370
|
ensemble.rmsf(models) # per-atom fluctuation
|
|
282
371
|
ensemble.average(models) # mean structure
|
|
@@ -324,7 +413,7 @@ to the common ML frameworks. The base `to_graph()` needs no extra dependencies;
|
|
|
324
413
|
each exporter imports its backend lazily.
|
|
325
414
|
|
|
326
415
|
```python
|
|
327
|
-
mol = ms.read("1fqy.pdb")
|
|
416
|
+
mol = ms.read("examples/data/1fqy.pdb")
|
|
328
417
|
|
|
329
418
|
g = mol.to_graph() # MolecularGraph: nodes + edges, no deps
|
|
330
419
|
g.n_atoms, g.n_bonds # counts
|
|
@@ -353,6 +442,18 @@ Graph feature presets are also available through
|
|
|
353
442
|
`mol.to_graph(include_chemical_features=True)` to attach optional RDKit-backed
|
|
354
443
|
aromatic atom and bond flags.
|
|
355
444
|
|
|
445
|
+
For protein-scale spatial graphs, build a residue contact graph instead:
|
|
446
|
+
|
|
447
|
+
```python
|
|
448
|
+
rg = mol.to_residue_contact_graph(cutoff=8.0, method="ca", min_seq_sep=4)
|
|
449
|
+
RG = rg.to_networkx()
|
|
450
|
+
residue_data = rg.to_pyg_data(node_preset="ml", edge_preset="ml")
|
|
451
|
+
```
|
|
452
|
+
|
|
453
|
+
Use atom/bond graphs when covalent chemistry is the signal. Use residue or
|
|
454
|
+
bead contact graphs when 3D neighborhoods, interfaces, folded shape or
|
|
455
|
+
long-range contacts are the signal.
|
|
456
|
+
|
|
356
457
|
### Coarse-graining
|
|
357
458
|
|
|
358
459
|
Map an atomistic structure onto a smaller set of beads. The result is an
|
|
@@ -360,11 +461,12 @@ ordinary `Molecule` (beads as "atoms") with explicit CG bonds attached, so it
|
|
|
360
461
|
plots, transforms and graphs like anything else.
|
|
361
462
|
|
|
362
463
|
```python
|
|
363
|
-
mol = ms.read("1fqy.pdb")
|
|
464
|
+
mol = ms.read("examples/data/1fqy.pdb")
|
|
364
465
|
|
|
365
466
|
cg = mol.coarse_grain("residue_com") # one bead per residue (centre of mass)
|
|
366
467
|
cg = mol.coarse_grain("residue_centroid") # ...or geometric centroid
|
|
367
468
|
cg = mol.coarse_grain("martini") # simplified backbone + side-chain beads
|
|
469
|
+
cg = mol.coarse_grain("martini", virtual_sites=[{"name": "MID", "parents": [0, 2]}])
|
|
368
470
|
cg.plot(scale=200) # beads + backbone topology
|
|
369
471
|
print(cg.mapping_report()) # explain beads, dropped atoms, and bonds
|
|
370
472
|
|
|
@@ -380,31 +482,68 @@ cg = mol.coarse_grain({"head": [0, 1, 2, 3], "tail": [4, 5, 6, 7]},
|
|
|
380
482
|
cg.to_graph() # CG bead network, ready for ML
|
|
381
483
|
```
|
|
382
484
|
|
|
485
|
+
Visualise the mapping, inspect the per-bead assignment, and export it:
|
|
486
|
+
|
|
487
|
+
```python
|
|
488
|
+
ms.plot_mapping(mol, cg) # atoms coloured by bead, beads + bonds overlaid
|
|
489
|
+
|
|
490
|
+
report = cg.coarse_grain_report # structured assignment
|
|
491
|
+
print(report.coverage()) # "426 beads from 1661/1661 atoms"
|
|
492
|
+
print(report.beads[0].atom_indices) # source atoms folded into bead 0
|
|
493
|
+
|
|
494
|
+
cg.write_mapping("mapping.json") # round-trippable JSON record
|
|
495
|
+
record = ms.read_cg_mapping("mapping.json")
|
|
496
|
+
cg2 = ms.apply_cg_mapping(mol, record) # rebuild on a matching structure
|
|
497
|
+
cg.write_index("mapping.ndx") # GROMACS-style index, one group per bead
|
|
498
|
+
```
|
|
499
|
+
|
|
383
500
|
Bead positions are mass-weighted (or centroids). For residue mappings bonds are
|
|
384
501
|
generated automatically (within a residue, plus a backbone chain between
|
|
385
502
|
residues); pass `bonds=` to define them yourself. Name-based bonds are intended
|
|
386
503
|
for unique bead names such as `head`/`tail`; repeated names such as `BB`/`SC`
|
|
387
504
|
are ambiguous, so use bead indices for those. Atoms you leave unassigned are
|
|
388
|
-
dropped with a warning.
|
|
389
|
-
|
|
390
|
-
Martini
|
|
505
|
+
dropped with a warning. The `"martini"` mapping teaches the idea of backbone and
|
|
506
|
+
sidechain beads. Virtual sites are represented explicitly as derived coordinate
|
|
507
|
+
sites and graph flags, but MolScope does not assign Martini bead types,
|
|
508
|
+
bonded/nonbonded parameters, charges, exclusions, GROMACS virtual-site topology
|
|
509
|
+
sections, or simulation-engine topology files. This is meant for teaching and
|
|
510
|
+
prototyping CG mappings, not as a replacement for production Martini
|
|
511
|
+
preparation: the JSON and `.ndx` exports describe a bead assignment for
|
|
512
|
+
inspection and reuse, not a validated simulation topology.
|
|
513
|
+
|
|
514
|
+
## Command-line interface
|
|
391
515
|
|
|
392
|
-
|
|
516
|
+
MolScope provides a powerful CLI for visualization, batch analysis, and ML graph export.
|
|
393
517
|
|
|
518
|
+
### View (default)
|
|
519
|
+
Visualize a structure, apply transformations, and save images or animations.
|
|
394
520
|
```bash
|
|
395
|
-
molscope
|
|
396
|
-
molscope 1fqy.pdb --select atom_name=CA --
|
|
521
|
+
molscope examples/data/1fqy.pdb --select atom_name=CA --color-by residue --save ca.png
|
|
522
|
+
molscope examples/data/1fqy.pdb --select "chain=A and atom_name=CA" --save chain-a-ca.png
|
|
397
523
|
molscope --fetch 1aml --center --gif amyloid.gif
|
|
398
|
-
python -m molscope 1fqy.pdb # equivalent if not pip-installed
|
|
399
524
|
```
|
|
400
525
|
|
|
526
|
+
### Analyze
|
|
527
|
+
Batch compute molecular descriptors for many files and save to a CSV table.
|
|
528
|
+
```bash
|
|
529
|
+
molscope analyze examples/data/*.pdb --out results.csv --preset native-3d --jobs 4
|
|
530
|
+
```
|
|
531
|
+
|
|
532
|
+
### Export
|
|
533
|
+
Batch export molecular graphs to PyTorch Geometric, DGL, or NetworkX formats.
|
|
534
|
+
```bash
|
|
535
|
+
molscope export "data/*.cif" --to pyg --out-dir pyg_graphs/ --pe laplacian --jobs 8
|
|
536
|
+
```
|
|
537
|
+
Supports advanced features like `--self-loops`, `--global-node`, and `--pe` (positional encodings).
|
|
538
|
+
|
|
539
|
+
|
|
401
540
|
## Sample structures
|
|
402
541
|
|
|
403
542
|
| File | Contents |
|
|
404
543
|
|------|----------|
|
|
405
|
-
| `helix_201.xyz` | a helix (bare coordinates) |
|
|
406
|
-
| `1fqy.pdb` | Aquaporin-1, single model (1661 atoms) |
|
|
407
|
-
| `1aml.pdb` | Alzheimer amyloid A4 peptide, 20-model NMR ensemble |
|
|
544
|
+
| `examples/data/helix_201.xyz` | a helix (bare coordinates) |
|
|
545
|
+
| `examples/data/1fqy.pdb` | Aquaporin-1, single model (1661 atoms) |
|
|
546
|
+
| `examples/data/1aml.pdb` | Alzheimer amyloid A4 peptide, 20-model NMR ensemble |
|
|
408
547
|
|
|
409
548
|
## Notes
|
|
410
549
|
|
|
@@ -422,6 +561,7 @@ python -m molscope 1fqy.pdb # equivalent if not pip-installed
|
|
|
422
561
|
- Optional extras: `pip install "molscope[fast]"` (scipy, faster bonds/contacts),
|
|
423
562
|
`"molscope[viz]"` (py3Dmol, for `Molecule.view`), `"molscope[graph]"`
|
|
424
563
|
(NetworkX), `"molscope[chem]"` (RDKit), `"molscope[cif]"` (Gemmi),
|
|
564
|
+
`"molscope[gpu]"` (PyTorch dense distance/contact-map backend),
|
|
425
565
|
`"molscope[pyg]"`, `"molscope[dgl]"`, or `"molscope[gnn]"`. For custom CUDA,
|
|
426
566
|
ROCm, Apple Silicon, or cluster builds, install the matching PyTorch stack
|
|
427
567
|
first.
|
|
@@ -429,11 +569,18 @@ python -m molscope 1fqy.pdb # equivalent if not pip-installed
|
|
|
429
569
|
## Tests and linting
|
|
430
570
|
|
|
431
571
|
```bash
|
|
432
|
-
uv run pytest
|
|
433
|
-
uv run
|
|
572
|
+
uv run pytest # full test suite
|
|
573
|
+
uv run pytest tests/validation # validation suite only
|
|
574
|
+
uv run ruff check . # lint
|
|
434
575
|
```
|
|
435
576
|
|
|
436
|
-
CI (GitHub Actions) runs
|
|
577
|
+
CI (GitHub Actions) runs the suite and linting across Python 3.9 / 3.11 / 3.13,
|
|
578
|
+
smoke-imports the optional extras, and runs a separate **validation** job on
|
|
579
|
+
every push and PR. `tests/validation` is a two-tier suite: dependency-free
|
|
580
|
+
physical invariants (rigid-body algebra, geometry, coarse-grain conservation)
|
|
581
|
+
that run everywhere, plus cross-checks against reference scientific tools (the
|
|
582
|
+
simplified DSSP vs `mkdssp`) that turn "the tests pass" into a measured
|
|
583
|
+
agreement number.
|
|
437
584
|
|
|
438
585
|
## License
|
|
439
586
|
|