molscope 0.6.2__tar.gz → 0.8.0__tar.gz
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
- {molscope-0.6.2 → molscope-0.8.0}/PKG-INFO +127 -22
- {molscope-0.6.2 → molscope-0.8.0}/README.md +111 -21
- {molscope-0.6.2 → molscope-0.8.0}/molscope/__init__.py +24 -6
- molscope-0.8.0/molscope/chem.py +204 -0
- molscope-0.8.0/molscope/cif.py +154 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/contactmap.py +5 -3
- {molscope-0.6.2 → molscope-0.8.0}/molscope/descriptors.py +202 -11
- molscope-0.8.0/molscope/dssp.py +232 -0
- molscope-0.8.0/molscope/graph.py +305 -0
- molscope-0.8.0/molscope/io.py +690 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/molecule.py +234 -19
- {molscope-0.6.2 → molscope-0.8.0}/molscope/plotting.py +7 -2
- {molscope-0.6.2 → molscope-0.8.0}/molscope.egg-info/PKG-INFO +127 -22
- {molscope-0.6.2 → molscope-0.8.0}/molscope.egg-info/SOURCES.txt +7 -0
- molscope-0.8.0/molscope.egg-info/requires.txt +34 -0
- {molscope-0.6.2 → molscope-0.8.0}/pyproject.toml +14 -2
- molscope-0.8.0/tests/test_chem.py +51 -0
- molscope-0.8.0/tests/test_cif_validation.py +69 -0
- molscope-0.8.0/tests/test_descriptors.py +159 -0
- molscope-0.8.0/tests/test_dssp.py +67 -0
- molscope-0.8.0/tests/test_extras.py +28 -0
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_features.py +73 -0
- molscope-0.8.0/tests/test_graph.py +193 -0
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_io.py +65 -1
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_molecule.py +24 -0
- molscope-0.6.2/molscope/graph.py +0 -151
- molscope-0.6.2/molscope/io.py +0 -342
- molscope-0.6.2/molscope.egg-info/requires.txt +0 -14
- molscope-0.6.2/tests/test_descriptors.py +0 -73
- molscope-0.6.2/tests/test_graph.py +0 -99
- {molscope-0.6.2 → molscope-0.8.0}/LICENSE +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/__main__.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/cli.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/coarsegrain.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/elements.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope/ensemble.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope.egg-info/dependency_links.txt +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope.egg-info/entry_points.txt +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/molscope.egg-info/top_level.txt +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/setup.cfg +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_clustering.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_coarsegrain.py +0 -0
- {molscope-0.6.2 → molscope-0.8.0}/tests/test_contactmap.py +0 -0
|
@@ -1,6 +1,6 @@
|
|
|
1
1
|
Metadata-Version: 2.4
|
|
2
2
|
Name: molscope
|
|
3
|
-
Version: 0.
|
|
3
|
+
Version: 0.8.0
|
|
4
4
|
Summary: Lightweight molecular structure analysis, visualisation, graph export, and coarse-graining in Python.
|
|
5
5
|
Author-email: Roshan Shrestha <roshanpra@gmail.com>
|
|
6
6
|
License-Expression: MIT
|
|
@@ -23,6 +23,21 @@ Provides-Extra: viz
|
|
|
23
23
|
Requires-Dist: py3Dmol>=2.0; extra == "viz"
|
|
24
24
|
Provides-Extra: graph
|
|
25
25
|
Requires-Dist: networkx>=2.6; extra == "graph"
|
|
26
|
+
Provides-Extra: chem
|
|
27
|
+
Requires-Dist: rdkit>=2023.9; extra == "chem"
|
|
28
|
+
Provides-Extra: cif
|
|
29
|
+
Requires-Dist: gemmi>=0.7; extra == "cif"
|
|
30
|
+
Provides-Extra: pyg
|
|
31
|
+
Requires-Dist: torch>=2.0; extra == "pyg"
|
|
32
|
+
Requires-Dist: torch-geometric>=2.3; extra == "pyg"
|
|
33
|
+
Provides-Extra: dgl
|
|
34
|
+
Requires-Dist: torch>=2.0; extra == "dgl"
|
|
35
|
+
Requires-Dist: dgl>=1.1; extra == "dgl"
|
|
36
|
+
Provides-Extra: gnn
|
|
37
|
+
Requires-Dist: networkx>=2.6; extra == "gnn"
|
|
38
|
+
Requires-Dist: torch>=2.0; extra == "gnn"
|
|
39
|
+
Requires-Dist: torch-geometric>=2.3; extra == "gnn"
|
|
40
|
+
Requires-Dist: dgl>=1.1; extra == "gnn"
|
|
26
41
|
Dynamic: license-file
|
|
27
42
|
|
|
28
43
|
# MolScope
|
|
@@ -35,29 +50,65 @@ Dynamic: license-file
|
|
|
35
50
|
Lightweight molecular structure analysis, visualisation, graph export, and
|
|
36
51
|
coarse-graining in Python. Read `.xyz`, `.pdb`, `.cif` and `.sdf` files
|
|
37
52
|
(optionally gzip-compressed), select and analyse atoms, and visualise them in
|
|
38
|
-
3D. The `.cif` reader
|
|
39
|
-
|
|
53
|
+
3D. The `.cif` reader handles standard `_atom_site` coordinate loops, including
|
|
54
|
+
quoted values; optional Gemmi-backed validation is available through
|
|
55
|
+
`pip install "molscope[cif]"`.
|
|
40
56
|
|
|
41
|
-
| 3D structure
|
|
42
|
-
| --- | --- | --- |
|
|
43
|
-
|  |  | 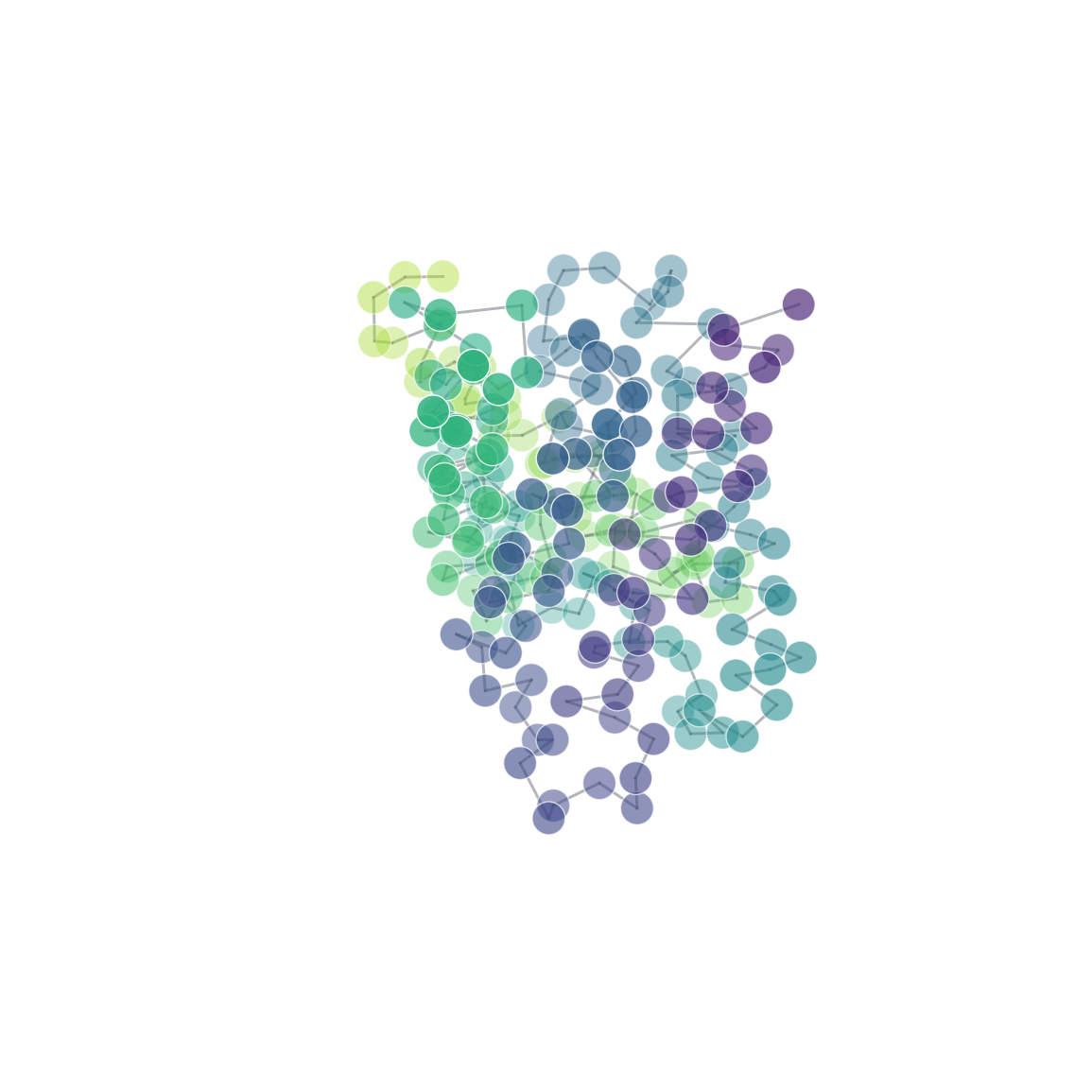 |
|
|
57
|
+
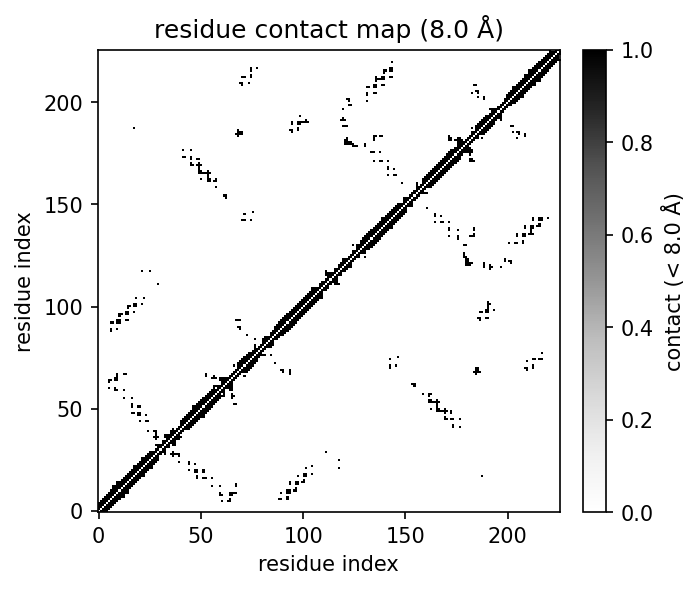
| 3D structure (element) | Secondary structure (DSSP) | Residue contact map | Coarse-grained beads |
|
|
58
|
+
| --- | --- | --- | --- |
|
|
59
|
+
|  | 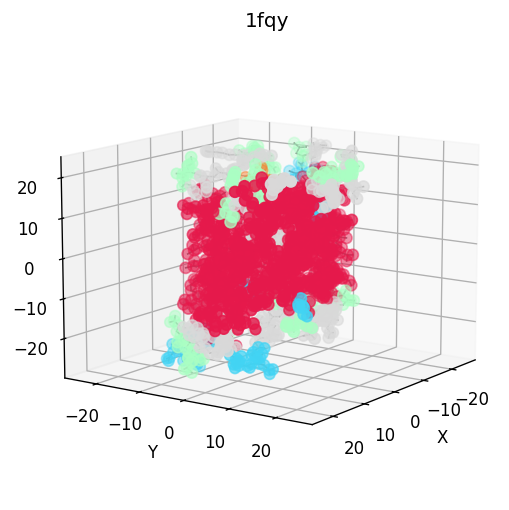 |  | 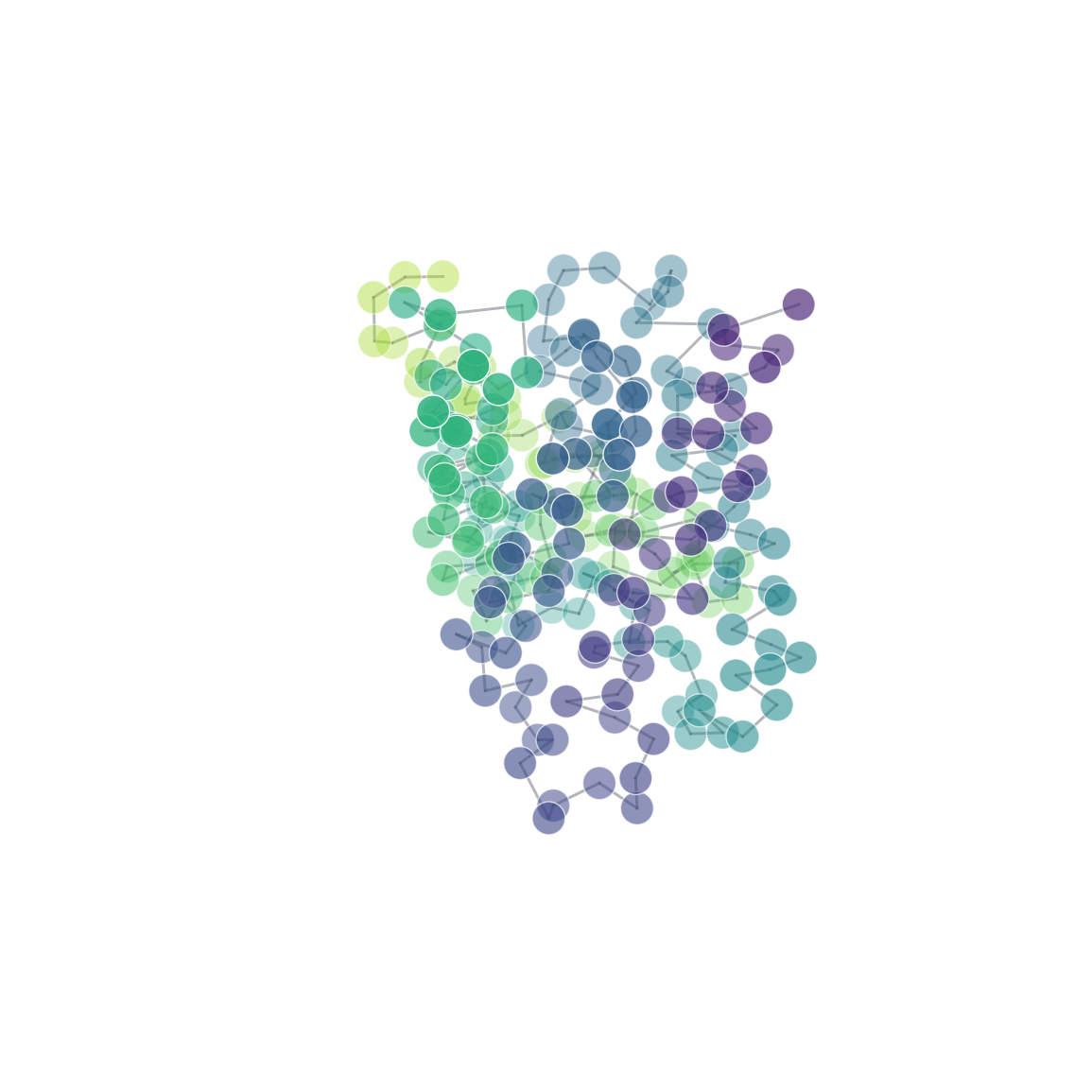 |
|
|
44
60
|
|
|
45
61
|
## What it does
|
|
46
62
|
|
|
47
|
-
- **Read and write** XYZ, PDB, mmCIF and SDF (gzip-aware),
|
|
48
|
-
|
|
63
|
+
- **Read and write** XYZ, PDB, mmCIF and SDF (gzip-aware), preserve SDF/PDB
|
|
64
|
+
explicit bonds and SDF formal charges where present, fetch structures by id
|
|
65
|
+
from RCSB, and load multi-model NMR ensembles.
|
|
66
|
+
- **Validate mmCIF** syntax, atom-site coordinate columns, and supplied
|
|
67
|
+
dictionary files with optional Gemmi support.
|
|
49
68
|
- **Select and measure** by chain, element or residue; compute distances,
|
|
50
69
|
angles, dihedrals and Kabsch-aligned RMSD.
|
|
51
|
-
- **Analyse** centroids, radius of gyration, the inertia tensor,
|
|
52
|
-
and contacts.
|
|
70
|
+
- **Analyse** centroids, radius of gyration, the inertia tensor,
|
|
71
|
+
explicit/inferred bonds, and contacts.
|
|
53
72
|
- **Contact maps** at atom or residue level, with heatmap plots.
|
|
73
|
+
- **Secondary structure** via a self-contained, dependency-free DSSP, with
|
|
74
|
+
`plot(color_by="ss")`.
|
|
54
75
|
- **Ensembles**: pairwise RMSD, RMSF, averaging, and conformer clustering.
|
|
55
76
|
- **Export for ML**: flat structural descriptors and molecular graphs for
|
|
56
77
|
NetworkX, PyTorch Geometric and DGL.
|
|
78
|
+
- **Chemical perception and descriptors**: optional RDKit-backed formal charge,
|
|
79
|
+
valence, aromaticity and scalar descriptor features with
|
|
80
|
+
`pip install "molscope[chem]"`.
|
|
57
81
|
- **Coarse-grain** onto residue, Martini-style or custom bead mappings.
|
|
58
82
|
- **Visualise** with 3D matplotlib plots, an interactive py3Dmol viewer, spin
|
|
59
83
|
GIFs, and a command-line interface.
|
|
60
84
|
|
|
85
|
+
## Why MolScope?
|
|
86
|
+
|
|
87
|
+
MolScope is **not** intended to replace full molecular-simulation or
|
|
88
|
+
cheminformatics frameworks. It is a lightweight **educational and prototyping**
|
|
89
|
+
toolkit for reading common molecular structure files, performing simple
|
|
90
|
+
structural analysis, exporting graph representations for ML workflows, and
|
|
91
|
+
experimenting with coarse-grained mappings. Its core depends only on NumPy and
|
|
92
|
+
Matplotlib, and the API is Python-first and scriptable.
|
|
93
|
+
|
|
94
|
+
In particular, the coarse-graining tools are for **educational CG mapping and
|
|
95
|
+
bead-graph prototyping**: useful for exploring mappings before moving to a
|
|
96
|
+
production Martini workflow. They are not a validated Martini force-field
|
|
97
|
+
generator.
|
|
98
|
+
|
|
99
|
+
| Tool | Main focus | How MolScope differs |
|
|
100
|
+
| --- | --- | --- |
|
|
101
|
+
| RDKit | Cheminformatics | MolScope leans toward structure visualisation, protein/PDB-style metadata, and CG prototyping |
|
|
102
|
+
| MDAnalysis | MD trajectories | MolScope is lighter and easier for static structures and teaching |
|
|
103
|
+
| MDTraj | Trajectory analysis | MolScope is simpler and graph/CG oriented |
|
|
104
|
+
| Biopython | Structure parsing / bioinformatics | MolScope adds 3D analysis, ML-graph export, and coarse-graining |
|
|
105
|
+
| PyMOL / VMD | Interactive visualisation | MolScope is Python-first, scriptable, and ML-export friendly |
|
|
106
|
+
| nglview | Notebook structure viewer | MolScope also does analysis, descriptors, graphs and CG, not just viewing |
|
|
107
|
+
|
|
108
|
+
Reach for those tools when you need their depth and validation. Reach for
|
|
109
|
+
MolScope when you want something small, readable, and quick to teach or
|
|
110
|
+
prototype with.
|
|
111
|
+
|
|
61
112
|
## Install
|
|
62
113
|
|
|
63
114
|
With [uv](https://docs.astral.sh/uv/) (recommended):
|
|
@@ -144,6 +195,7 @@ mol.radius_of_gyration # compactness (angstrom)
|
|
|
144
195
|
mol.dimensions, mol.formula # bounding box, Hill-order formula
|
|
145
196
|
mol.bonds() # inferred bond index pairs (KD-tree if scipy)
|
|
146
197
|
mol.contacts(cutoff=5.0) # atom pairs within a distance
|
|
198
|
+
mol.contact_count(cutoff=5.0) # count pairs without returning them
|
|
147
199
|
|
|
148
200
|
mol.distance(i, j) # bond length
|
|
149
201
|
mol.angle(i, j, k) # bond angle (degrees)
|
|
@@ -156,12 +208,14 @@ a.alpha_carbons().rmsd(b.alpha_carbons(), align=True) # CA-RMSD after Kabsch f
|
|
|
156
208
|
|
|
157
209
|
```python
|
|
158
210
|
features = mol.descriptors() # flat dict of scalar/vector descriptors
|
|
211
|
+
features = mol.descriptors(preset="native-3d")
|
|
159
212
|
features["radius_of_gyration"]
|
|
160
213
|
features["principal_moments"] # 3 values
|
|
161
214
|
features["distance_histogram"] # fixed-size histogram
|
|
162
215
|
|
|
163
216
|
X, names = ms.featurize_many(
|
|
164
217
|
["a.pdb", "b.pdb", "c.xyz"],
|
|
218
|
+
preset="native-basic",
|
|
165
219
|
return_names=True,
|
|
166
220
|
) # numeric matrix + column names
|
|
167
221
|
```
|
|
@@ -170,7 +224,17 @@ Descriptors include atom/residue counts, element counts, molecular mass,
|
|
|
170
224
|
centres, radius of gyration, bounding-box dimensions, inertia tensor, principal
|
|
171
225
|
moments/axes, shape anisotropy, compactness, distance histograms, bond-length
|
|
172
226
|
summary statistics, and atom/residue contact summaries. Full contact maps remain
|
|
173
|
-
available through `mol.contact_map(...)`.
|
|
227
|
+
available through `mol.contact_map(...)`. With `pip install "molscope[chem]"`,
|
|
228
|
+
you can also request RDKit descriptors directly:
|
|
229
|
+
|
|
230
|
+
```python
|
|
231
|
+
mol.rdkit_descriptors(names=["MolWt", "TPSA"])
|
|
232
|
+
mol.descriptors(include_rdkit=True, rdkit_descriptor_names=["MolWt", "TPSA"])
|
|
233
|
+
```
|
|
234
|
+
|
|
235
|
+
For reproducible ML columns, use descriptor presets: `native-basic`,
|
|
236
|
+
`native-3d`, or `rdkit-basic`. Inspect the flattened column order with
|
|
237
|
+
`ms.descriptor_feature_names(...)`.
|
|
174
238
|
|
|
175
239
|
### Contact maps
|
|
176
240
|
|
|
@@ -184,6 +248,29 @@ mol.contact_map(level="residue", method="min") # closest inter-residue at
|
|
|
184
248
|
mol.contact_map(level="residue", method="com") # residue centre of mass
|
|
185
249
|
```
|
|
186
250
|
|
|
251
|
+
### Secondary structure (DSSP)
|
|
252
|
+
|
|
253
|
+
Assign protein secondary structure from backbone hydrogen-bond patterns with a
|
|
254
|
+
self-contained, pure-NumPy DSSP (no external `mkdssp` binary needed):
|
|
255
|
+
|
|
256
|
+
```python
|
|
257
|
+
mol = ms.read("1fqy.pdb")
|
|
258
|
+
ss = mol.secondary_structure() # SecondaryStructure, one code per residue
|
|
259
|
+
|
|
260
|
+
ss.string # e.g. '--HHHHHHHH--SS--EEEE--'
|
|
261
|
+
ss.codes # per-residue array
|
|
262
|
+
ss.summary() # helix/strand/coil counts and fractions
|
|
263
|
+
|
|
264
|
+
mol.plot(color_by="ss") # colour the 3D view by secondary structure
|
|
265
|
+
```
|
|
266
|
+
|
|
267
|
+
Codes follow DSSP: `H`/`G`/`I` helices, `E`/`B` strands, `T` turn, `S` bend,
|
|
268
|
+
`-` coil. This is a simplified **educational** implementation: it reproduces the
|
|
269
|
+
main classes from the Kabsch-Sander hydrogen-bond model but is not bit-identical
|
|
270
|
+
to the reference `mkdssp` on every edge case. It needs backbone N/CA/C/O atoms,
|
|
271
|
+
so use PDB/mmCIF input (not a bare `.xyz`). The secondary-structure render in the
|
|
272
|
+
showcase above (helices red, turns cyan, coil grey) is produced this way.
|
|
273
|
+
|
|
187
274
|
### NMR ensembles
|
|
188
275
|
|
|
189
276
|
```python
|
|
@@ -232,9 +319,9 @@ spin_gif(mol, "spin.gif") # rotating animation
|
|
|
232
319
|
|
|
233
320
|
### Molecular graphs (for machine learning)
|
|
234
321
|
|
|
235
|
-
Turn 3D coordinates plus inferred bonds into a graph, then export
|
|
236
|
-
ML frameworks. The base `to_graph()` needs no extra dependencies;
|
|
237
|
-
imports its backend lazily.
|
|
322
|
+
Turn 3D coordinates plus explicit or inferred bonds into a graph, then export
|
|
323
|
+
to the common ML frameworks. The base `to_graph()` needs no extra dependencies;
|
|
324
|
+
each exporter imports its backend lazily.
|
|
238
325
|
|
|
239
326
|
```python
|
|
240
327
|
mol = ms.read("1fqy.pdb")
|
|
@@ -243,18 +330,28 @@ g = mol.to_graph() # MolecularGraph: nodes + edges, no deps
|
|
|
243
330
|
g.n_atoms, g.n_bonds # counts
|
|
244
331
|
g.atomic_numbers, g.masses # per-node arrays
|
|
245
332
|
g.node_features() # (N, 2) default features [atomic_number, mass]
|
|
333
|
+
g.node_features("ml") # stable ML node preset
|
|
334
|
+
g.edge_features("ml") # stable ML edge preset
|
|
246
335
|
|
|
247
336
|
G = mol.to_networkx() # networkx.Graph with node/edge attributes
|
|
248
337
|
data = mol.to_pyg_data() # torch_geometric.data.Data (x, pos, edge_index, edge_attr, z)
|
|
249
338
|
dglg = mol.to_dgl_graph() # dgl.DGLGraph with ndata/edata tensors
|
|
250
339
|
```
|
|
251
340
|
|
|
252
|
-
Nodes carry element, atomic number, mass, coordinates and (from
|
|
253
|
-
name, residue and chain. Edges carry the bonded pair,
|
|
254
|
-
bond order (`1.0` for
|
|
255
|
-
|
|
256
|
-
|
|
257
|
-
`
|
|
341
|
+
Nodes carry element, atomic number, mass, coordinates, formal charge, and (from
|
|
342
|
+
PDB/mmCIF) atom name, residue and chain. Edges carry the bonded pair,
|
|
343
|
+
interatomic distance, and bond order from SDF where available (`1.0` for
|
|
344
|
+
PDB/CONECT or geometrically inferred bonds). Install backends as needed:
|
|
345
|
+
`pip install "molscope[graph]"` for NetworkX, `"molscope[pyg]"` for PyTorch
|
|
346
|
+
Geometric, `"molscope[dgl]"` for DGL, or `"molscope[gnn]"` for all graph
|
|
347
|
+
backends. For custom CUDA, ROCm, Apple Silicon, or cluster builds, install the
|
|
348
|
+
matching PyTorch stack first.
|
|
349
|
+
|
|
350
|
+
Graph feature presets are also available through
|
|
351
|
+
`mol.to_pyg_data(node_preset="ml", edge_preset="ml")` and
|
|
352
|
+
`mol.to_dgl_graph(node_preset="ml", edge_preset="ml")`. Use
|
|
353
|
+
`mol.to_graph(include_chemical_features=True)` to attach optional RDKit-backed
|
|
354
|
+
aromatic atom and bond flags.
|
|
258
355
|
|
|
259
356
|
### Coarse-graining
|
|
260
357
|
|
|
@@ -314,12 +411,20 @@ python -m molscope 1fqy.pdb # equivalent if not pip-installed
|
|
|
314
411
|
- PDB files are parsed by **fixed columns**, not whitespace splitting, so atoms
|
|
315
412
|
with touching coordinate fields (large or negative values) read correctly.
|
|
316
413
|
- Alternate conformations (altLoc) other than the primary one are skipped.
|
|
414
|
+
Use `read_pdb(..., altloc="first"|"highest_occupancy"|"all")` to select a
|
|
415
|
+
different policy.
|
|
317
416
|
- `read_pdb` returns a single model (`model=1` by default); use `read_pdb_models`
|
|
318
417
|
for the whole ensemble.
|
|
418
|
+
- SDF/MOL V2000 bond blocks, formal charges, and PDB `CONECT` records are
|
|
419
|
+
preserved. PDB output writes explicit bonds back as `CONECT` records.
|
|
319
420
|
- Bond inference uses a `scipy.spatial.cKDTree` when available; without scipy it
|
|
320
421
|
falls back to a dense `O(n^2)` search that is refused above ~8000 atoms.
|
|
321
|
-
- Optional extras: `pip install "molscope[fast]"` (scipy, faster bonds/contacts)
|
|
322
|
-
|
|
422
|
+
- Optional extras: `pip install "molscope[fast]"` (scipy, faster bonds/contacts),
|
|
423
|
+
`"molscope[viz]"` (py3Dmol, for `Molecule.view`), `"molscope[graph]"`
|
|
424
|
+
(NetworkX), `"molscope[chem]"` (RDKit), `"molscope[cif]"` (Gemmi),
|
|
425
|
+
`"molscope[pyg]"`, `"molscope[dgl]"`, or `"molscope[gnn]"`. For custom CUDA,
|
|
426
|
+
ROCm, Apple Silicon, or cluster builds, install the matching PyTorch stack
|
|
427
|
+
first.
|
|
323
428
|
|
|
324
429
|
## Tests and linting
|
|
325
430
|
|
|
@@ -8,29 +8,65 @@
|
|
|
8
8
|
Lightweight molecular structure analysis, visualisation, graph export, and
|
|
9
9
|
coarse-graining in Python. Read `.xyz`, `.pdb`, `.cif` and `.sdf` files
|
|
10
10
|
(optionally gzip-compressed), select and analyse atoms, and visualise them in
|
|
11
|
-
3D. The `.cif` reader
|
|
12
|
-
|
|
11
|
+
3D. The `.cif` reader handles standard `_atom_site` coordinate loops, including
|
|
12
|
+
quoted values; optional Gemmi-backed validation is available through
|
|
13
|
+
`pip install "molscope[cif]"`.
|
|
13
14
|
|
|
14
|
-
| 3D structure
|
|
15
|
-
| --- | --- | --- |
|
|
16
|
-
|  |  |  |
|
|
15
|
+
| 3D structure (element) | Secondary structure (DSSP) | Residue contact map | Coarse-grained beads |
|
|
16
|
+
| --- | --- | --- | --- |
|
|
17
|
+
|  |  |  |  |
|
|
17
18
|
|
|
18
19
|
## What it does
|
|
19
20
|
|
|
20
|
-
- **Read and write** XYZ, PDB, mmCIF and SDF (gzip-aware),
|
|
21
|
-
|
|
21
|
+
- **Read and write** XYZ, PDB, mmCIF and SDF (gzip-aware), preserve SDF/PDB
|
|
22
|
+
explicit bonds and SDF formal charges where present, fetch structures by id
|
|
23
|
+
from RCSB, and load multi-model NMR ensembles.
|
|
24
|
+
- **Validate mmCIF** syntax, atom-site coordinate columns, and supplied
|
|
25
|
+
dictionary files with optional Gemmi support.
|
|
22
26
|
- **Select and measure** by chain, element or residue; compute distances,
|
|
23
27
|
angles, dihedrals and Kabsch-aligned RMSD.
|
|
24
|
-
- **Analyse** centroids, radius of gyration, the inertia tensor,
|
|
25
|
-
and contacts.
|
|
28
|
+
- **Analyse** centroids, radius of gyration, the inertia tensor,
|
|
29
|
+
explicit/inferred bonds, and contacts.
|
|
26
30
|
- **Contact maps** at atom or residue level, with heatmap plots.
|
|
31
|
+
- **Secondary structure** via a self-contained, dependency-free DSSP, with
|
|
32
|
+
`plot(color_by="ss")`.
|
|
27
33
|
- **Ensembles**: pairwise RMSD, RMSF, averaging, and conformer clustering.
|
|
28
34
|
- **Export for ML**: flat structural descriptors and molecular graphs for
|
|
29
35
|
NetworkX, PyTorch Geometric and DGL.
|
|
36
|
+
- **Chemical perception and descriptors**: optional RDKit-backed formal charge,
|
|
37
|
+
valence, aromaticity and scalar descriptor features with
|
|
38
|
+
`pip install "molscope[chem]"`.
|
|
30
39
|
- **Coarse-grain** onto residue, Martini-style or custom bead mappings.
|
|
31
40
|
- **Visualise** with 3D matplotlib plots, an interactive py3Dmol viewer, spin
|
|
32
41
|
GIFs, and a command-line interface.
|
|
33
42
|
|
|
43
|
+
## Why MolScope?
|
|
44
|
+
|
|
45
|
+
MolScope is **not** intended to replace full molecular-simulation or
|
|
46
|
+
cheminformatics frameworks. It is a lightweight **educational and prototyping**
|
|
47
|
+
toolkit for reading common molecular structure files, performing simple
|
|
48
|
+
structural analysis, exporting graph representations for ML workflows, and
|
|
49
|
+
experimenting with coarse-grained mappings. Its core depends only on NumPy and
|
|
50
|
+
Matplotlib, and the API is Python-first and scriptable.
|
|
51
|
+
|
|
52
|
+
In particular, the coarse-graining tools are for **educational CG mapping and
|
|
53
|
+
bead-graph prototyping**: useful for exploring mappings before moving to a
|
|
54
|
+
production Martini workflow. They are not a validated Martini force-field
|
|
55
|
+
generator.
|
|
56
|
+
|
|
57
|
+
| Tool | Main focus | How MolScope differs |
|
|
58
|
+
| --- | --- | --- |
|
|
59
|
+
| RDKit | Cheminformatics | MolScope leans toward structure visualisation, protein/PDB-style metadata, and CG prototyping |
|
|
60
|
+
| MDAnalysis | MD trajectories | MolScope is lighter and easier for static structures and teaching |
|
|
61
|
+
| MDTraj | Trajectory analysis | MolScope is simpler and graph/CG oriented |
|
|
62
|
+
| Biopython | Structure parsing / bioinformatics | MolScope adds 3D analysis, ML-graph export, and coarse-graining |
|
|
63
|
+
| PyMOL / VMD | Interactive visualisation | MolScope is Python-first, scriptable, and ML-export friendly |
|
|
64
|
+
| nglview | Notebook structure viewer | MolScope also does analysis, descriptors, graphs and CG, not just viewing |
|
|
65
|
+
|
|
66
|
+
Reach for those tools when you need their depth and validation. Reach for
|
|
67
|
+
MolScope when you want something small, readable, and quick to teach or
|
|
68
|
+
prototype with.
|
|
69
|
+
|
|
34
70
|
## Install
|
|
35
71
|
|
|
36
72
|
With [uv](https://docs.astral.sh/uv/) (recommended):
|
|
@@ -117,6 +153,7 @@ mol.radius_of_gyration # compactness (angstrom)
|
|
|
117
153
|
mol.dimensions, mol.formula # bounding box, Hill-order formula
|
|
118
154
|
mol.bonds() # inferred bond index pairs (KD-tree if scipy)
|
|
119
155
|
mol.contacts(cutoff=5.0) # atom pairs within a distance
|
|
156
|
+
mol.contact_count(cutoff=5.0) # count pairs without returning them
|
|
120
157
|
|
|
121
158
|
mol.distance(i, j) # bond length
|
|
122
159
|
mol.angle(i, j, k) # bond angle (degrees)
|
|
@@ -129,12 +166,14 @@ a.alpha_carbons().rmsd(b.alpha_carbons(), align=True) # CA-RMSD after Kabsch f
|
|
|
129
166
|
|
|
130
167
|
```python
|
|
131
168
|
features = mol.descriptors() # flat dict of scalar/vector descriptors
|
|
169
|
+
features = mol.descriptors(preset="native-3d")
|
|
132
170
|
features["radius_of_gyration"]
|
|
133
171
|
features["principal_moments"] # 3 values
|
|
134
172
|
features["distance_histogram"] # fixed-size histogram
|
|
135
173
|
|
|
136
174
|
X, names = ms.featurize_many(
|
|
137
175
|
["a.pdb", "b.pdb", "c.xyz"],
|
|
176
|
+
preset="native-basic",
|
|
138
177
|
return_names=True,
|
|
139
178
|
) # numeric matrix + column names
|
|
140
179
|
```
|
|
@@ -143,7 +182,17 @@ Descriptors include atom/residue counts, element counts, molecular mass,
|
|
|
143
182
|
centres, radius of gyration, bounding-box dimensions, inertia tensor, principal
|
|
144
183
|
moments/axes, shape anisotropy, compactness, distance histograms, bond-length
|
|
145
184
|
summary statistics, and atom/residue contact summaries. Full contact maps remain
|
|
146
|
-
available through `mol.contact_map(...)`.
|
|
185
|
+
available through `mol.contact_map(...)`. With `pip install "molscope[chem]"`,
|
|
186
|
+
you can also request RDKit descriptors directly:
|
|
187
|
+
|
|
188
|
+
```python
|
|
189
|
+
mol.rdkit_descriptors(names=["MolWt", "TPSA"])
|
|
190
|
+
mol.descriptors(include_rdkit=True, rdkit_descriptor_names=["MolWt", "TPSA"])
|
|
191
|
+
```
|
|
192
|
+
|
|
193
|
+
For reproducible ML columns, use descriptor presets: `native-basic`,
|
|
194
|
+
`native-3d`, or `rdkit-basic`. Inspect the flattened column order with
|
|
195
|
+
`ms.descriptor_feature_names(...)`.
|
|
147
196
|
|
|
148
197
|
### Contact maps
|
|
149
198
|
|
|
@@ -157,6 +206,29 @@ mol.contact_map(level="residue", method="min") # closest inter-residue at
|
|
|
157
206
|
mol.contact_map(level="residue", method="com") # residue centre of mass
|
|
158
207
|
```
|
|
159
208
|
|
|
209
|
+
### Secondary structure (DSSP)
|
|
210
|
+
|
|
211
|
+
Assign protein secondary structure from backbone hydrogen-bond patterns with a
|
|
212
|
+
self-contained, pure-NumPy DSSP (no external `mkdssp` binary needed):
|
|
213
|
+
|
|
214
|
+
```python
|
|
215
|
+
mol = ms.read("1fqy.pdb")
|
|
216
|
+
ss = mol.secondary_structure() # SecondaryStructure, one code per residue
|
|
217
|
+
|
|
218
|
+
ss.string # e.g. '--HHHHHHHH--SS--EEEE--'
|
|
219
|
+
ss.codes # per-residue array
|
|
220
|
+
ss.summary() # helix/strand/coil counts and fractions
|
|
221
|
+
|
|
222
|
+
mol.plot(color_by="ss") # colour the 3D view by secondary structure
|
|
223
|
+
```
|
|
224
|
+
|
|
225
|
+
Codes follow DSSP: `H`/`G`/`I` helices, `E`/`B` strands, `T` turn, `S` bend,
|
|
226
|
+
`-` coil. This is a simplified **educational** implementation: it reproduces the
|
|
227
|
+
main classes from the Kabsch-Sander hydrogen-bond model but is not bit-identical
|
|
228
|
+
to the reference `mkdssp` on every edge case. It needs backbone N/CA/C/O atoms,
|
|
229
|
+
so use PDB/mmCIF input (not a bare `.xyz`). The secondary-structure render in the
|
|
230
|
+
showcase above (helices red, turns cyan, coil grey) is produced this way.
|
|
231
|
+
|
|
160
232
|
### NMR ensembles
|
|
161
233
|
|
|
162
234
|
```python
|
|
@@ -205,9 +277,9 @@ spin_gif(mol, "spin.gif") # rotating animation
|
|
|
205
277
|
|
|
206
278
|
### Molecular graphs (for machine learning)
|
|
207
279
|
|
|
208
|
-
Turn 3D coordinates plus inferred bonds into a graph, then export
|
|
209
|
-
ML frameworks. The base `to_graph()` needs no extra dependencies;
|
|
210
|
-
imports its backend lazily.
|
|
280
|
+
Turn 3D coordinates plus explicit or inferred bonds into a graph, then export
|
|
281
|
+
to the common ML frameworks. The base `to_graph()` needs no extra dependencies;
|
|
282
|
+
each exporter imports its backend lazily.
|
|
211
283
|
|
|
212
284
|
```python
|
|
213
285
|
mol = ms.read("1fqy.pdb")
|
|
@@ -216,18 +288,28 @@ g = mol.to_graph() # MolecularGraph: nodes + edges, no deps
|
|
|
216
288
|
g.n_atoms, g.n_bonds # counts
|
|
217
289
|
g.atomic_numbers, g.masses # per-node arrays
|
|
218
290
|
g.node_features() # (N, 2) default features [atomic_number, mass]
|
|
291
|
+
g.node_features("ml") # stable ML node preset
|
|
292
|
+
g.edge_features("ml") # stable ML edge preset
|
|
219
293
|
|
|
220
294
|
G = mol.to_networkx() # networkx.Graph with node/edge attributes
|
|
221
295
|
data = mol.to_pyg_data() # torch_geometric.data.Data (x, pos, edge_index, edge_attr, z)
|
|
222
296
|
dglg = mol.to_dgl_graph() # dgl.DGLGraph with ndata/edata tensors
|
|
223
297
|
```
|
|
224
298
|
|
|
225
|
-
Nodes carry element, atomic number, mass, coordinates and (from
|
|
226
|
-
name, residue and chain. Edges carry the bonded pair,
|
|
227
|
-
bond order (`1.0` for
|
|
228
|
-
|
|
229
|
-
|
|
230
|
-
`
|
|
299
|
+
Nodes carry element, atomic number, mass, coordinates, formal charge, and (from
|
|
300
|
+
PDB/mmCIF) atom name, residue and chain. Edges carry the bonded pair,
|
|
301
|
+
interatomic distance, and bond order from SDF where available (`1.0` for
|
|
302
|
+
PDB/CONECT or geometrically inferred bonds). Install backends as needed:
|
|
303
|
+
`pip install "molscope[graph]"` for NetworkX, `"molscope[pyg]"` for PyTorch
|
|
304
|
+
Geometric, `"molscope[dgl]"` for DGL, or `"molscope[gnn]"` for all graph
|
|
305
|
+
backends. For custom CUDA, ROCm, Apple Silicon, or cluster builds, install the
|
|
306
|
+
matching PyTorch stack first.
|
|
307
|
+
|
|
308
|
+
Graph feature presets are also available through
|
|
309
|
+
`mol.to_pyg_data(node_preset="ml", edge_preset="ml")` and
|
|
310
|
+
`mol.to_dgl_graph(node_preset="ml", edge_preset="ml")`. Use
|
|
311
|
+
`mol.to_graph(include_chemical_features=True)` to attach optional RDKit-backed
|
|
312
|
+
aromatic atom and bond flags.
|
|
231
313
|
|
|
232
314
|
### Coarse-graining
|
|
233
315
|
|
|
@@ -287,12 +369,20 @@ python -m molscope 1fqy.pdb # equivalent if not pip-installed
|
|
|
287
369
|
- PDB files are parsed by **fixed columns**, not whitespace splitting, so atoms
|
|
288
370
|
with touching coordinate fields (large or negative values) read correctly.
|
|
289
371
|
- Alternate conformations (altLoc) other than the primary one are skipped.
|
|
372
|
+
Use `read_pdb(..., altloc="first"|"highest_occupancy"|"all")` to select a
|
|
373
|
+
different policy.
|
|
290
374
|
- `read_pdb` returns a single model (`model=1` by default); use `read_pdb_models`
|
|
291
375
|
for the whole ensemble.
|
|
376
|
+
- SDF/MOL V2000 bond blocks, formal charges, and PDB `CONECT` records are
|
|
377
|
+
preserved. PDB output writes explicit bonds back as `CONECT` records.
|
|
292
378
|
- Bond inference uses a `scipy.spatial.cKDTree` when available; without scipy it
|
|
293
379
|
falls back to a dense `O(n^2)` search that is refused above ~8000 atoms.
|
|
294
|
-
- Optional extras: `pip install "molscope[fast]"` (scipy, faster bonds/contacts)
|
|
295
|
-
|
|
380
|
+
- Optional extras: `pip install "molscope[fast]"` (scipy, faster bonds/contacts),
|
|
381
|
+
`"molscope[viz]"` (py3Dmol, for `Molecule.view`), `"molscope[graph]"`
|
|
382
|
+
(NetworkX), `"molscope[chem]"` (RDKit), `"molscope[cif]"` (Gemmi),
|
|
383
|
+
`"molscope[pyg]"`, `"molscope[dgl]"`, or `"molscope[gnn]"`. For custom CUDA,
|
|
384
|
+
ROCm, Apple Silicon, or cluster builds, install the matching PyTorch stack
|
|
385
|
+
first.
|
|
296
386
|
|
|
297
387
|
## Tests and linting
|
|
298
388
|
|
|
@@ -7,8 +7,10 @@ beads, and visualise everything in 3D.
|
|
|
7
7
|
|
|
8
8
|
What it does
|
|
9
9
|
------------
|
|
10
|
-
- **Read and write** XYZ, PDB, mmCIF and SDF;
|
|
11
|
-
multi-model NMR ensembles
|
|
10
|
+
- **Read and write** XYZ, PDB, mmCIF and SDF; preserve SDF/PDB explicit bonds
|
|
11
|
+
and SDF formal charges; fetch by id from RCSB; load multi-model NMR ensembles;
|
|
12
|
+
validate CIF/mmCIF with optional Gemmi support
|
|
13
|
+
(:func:`read`, :func:`fetch`, :func:`read_pdb_models`, :func:`validate_cif`).
|
|
12
14
|
- **Select and measure** by chain, element or residue; distances, angles,
|
|
13
15
|
dihedrals and Kabsch-aligned RMSD (:class:`Molecule`).
|
|
14
16
|
- **Analyse** centroids, radius of gyration, inertia tensor, bonds and contacts.
|
|
@@ -17,6 +19,8 @@ What it does
|
|
|
17
19
|
(:mod:`molscope.ensemble`, :func:`cluster`, :func:`rmsd_matrix`).
|
|
18
20
|
- **Export for ML**: structural descriptors and molecular graphs for NetworkX,
|
|
19
21
|
PyTorch Geometric and DGL (:func:`descriptors`, :class:`MolecularGraph`).
|
|
22
|
+
- **Chemical perception**: optional RDKit-backed valence, aromaticity, charge
|
|
23
|
+
features and descriptors (:func:`chemical_features`, :func:`rdkit_descriptors`).
|
|
20
24
|
- **Coarse-grain** onto residue, Martini-style or custom bead mappings
|
|
21
25
|
(:mod:`molscope.coarsegrain`).
|
|
22
26
|
- **Visualise** with 3D matplotlib plots, an interactive py3Dmol viewer, and
|
|
@@ -37,13 +41,16 @@ Examples
|
|
|
37
41
|
See https://github.com/roshan2004/molscope for the full documentation.
|
|
38
42
|
"""
|
|
39
43
|
|
|
40
|
-
from . import coarsegrain, ensemble
|
|
44
|
+
from . import coarsegrain, dssp, ensemble
|
|
45
|
+
from .chem import ChemicalFeatures, chemical_features, rdkit_descriptors, to_rdkit
|
|
46
|
+
from .cif import CifValidationReport, validate_cif
|
|
41
47
|
from .coarsegrain import BeadMapping, BondMapping, CoarseGrainReport, DroppedAtom
|
|
42
48
|
from .contactmap import ContactMap
|
|
43
|
-
from .descriptors import descriptors, featurize_many
|
|
49
|
+
from .descriptors import descriptor_feature_names, descriptors, featurize_many
|
|
50
|
+
from .dssp import SecondaryStructure
|
|
44
51
|
from .ensemble import Clustering, cluster, rmsd_matrix
|
|
45
52
|
from .ensemble import contact_frequency as ensemble_contact_frequency
|
|
46
|
-
from .graph import MolecularGraph
|
|
53
|
+
from .graph import MolecularGraph, edge_feature_names, node_feature_names
|
|
47
54
|
from .io import (
|
|
48
55
|
fetch,
|
|
49
56
|
read,
|
|
@@ -61,6 +68,8 @@ from .plotting import plot_rmsd_heatmap
|
|
|
61
68
|
|
|
62
69
|
__all__ = [
|
|
63
70
|
"Clustering",
|
|
71
|
+
"ChemicalFeatures",
|
|
72
|
+
"CifValidationReport",
|
|
64
73
|
"BeadMapping",
|
|
65
74
|
"BondMapping",
|
|
66
75
|
"CoarseGrainReport",
|
|
@@ -68,11 +77,16 @@ __all__ = [
|
|
|
68
77
|
"DroppedAtom",
|
|
69
78
|
"Molecule",
|
|
70
79
|
"MolecularGraph",
|
|
80
|
+
"SecondaryStructure",
|
|
71
81
|
"cluster",
|
|
82
|
+
"chemical_features",
|
|
72
83
|
"coarsegrain",
|
|
84
|
+
"descriptor_feature_names",
|
|
73
85
|
"descriptors",
|
|
86
|
+
"dssp",
|
|
74
87
|
"ensemble",
|
|
75
88
|
"ensemble_contact_frequency",
|
|
89
|
+
"edge_feature_names",
|
|
76
90
|
"featurize_many",
|
|
77
91
|
"fetch",
|
|
78
92
|
"plot_rmsd_heatmap",
|
|
@@ -83,8 +97,12 @@ __all__ = [
|
|
|
83
97
|
"read_sdf",
|
|
84
98
|
"read_xyz",
|
|
85
99
|
"read_xyz_frames",
|
|
100
|
+
"rdkit_descriptors",
|
|
86
101
|
"rmsd_matrix",
|
|
102
|
+
"node_feature_names",
|
|
103
|
+
"to_rdkit",
|
|
104
|
+
"validate_cif",
|
|
87
105
|
"write_pdb",
|
|
88
106
|
"write_xyz",
|
|
89
107
|
]
|
|
90
|
-
__version__ = "0.
|
|
108
|
+
__version__ = "0.8.0"
|