biopipen 0.27.2__tar.gz → 0.27.4__tar.gz
This diff represents the content of publicly available package versions that have been released to one of the supported registries. The information contained in this diff is provided for informational purposes only and reflects changes between package versions as they appear in their respective public registries.
Potentially problematic release.
This version of biopipen might be problematic. Click here for more details.
- {biopipen-0.27.2 → biopipen-0.27.4}/PKG-INFO +2 -2
- biopipen-0.27.4/biopipen/__init__.py +1 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/delim.py +1 -1
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/plot.py +36 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/scrna.py +17 -9
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/scrna_metabolic_landscape.py +3 -3
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/snp.py +65 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/tcr.py +6 -6
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/delim/SampleInfo.R +6 -6
- biopipen-0.27.4/biopipen/scripts/plot/ROC.R +88 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/MarkersFinder.R +30 -5
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/MetaMarkers.R +17 -3
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ScFGSEA.R +21 -4
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratPreparing.R +113 -1
- biopipen-0.27.4/biopipen/scripts/snp/MatrixEQTL.R +157 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/pyproject.toml +2 -2
- {biopipen-0.27.2 → biopipen-0.27.4}/setup.py +2 -2
- biopipen-0.27.2/biopipen/__init__.py +0 -1
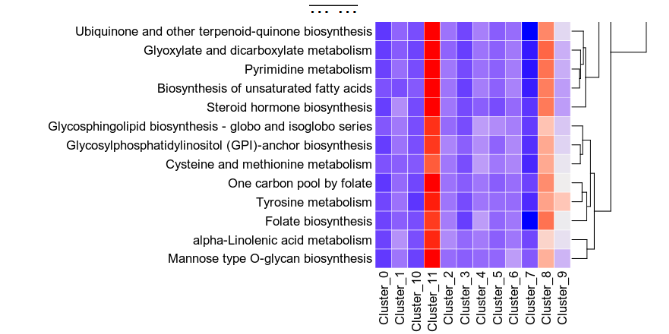
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/__init__.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/config.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/config.toml +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/defaults.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/filters.py +0 -0
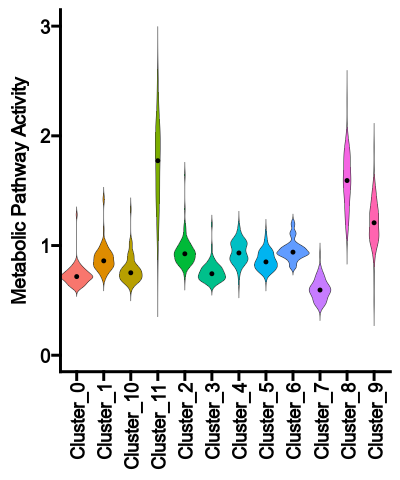
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/proc.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/core/testing.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/__init__.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/bam.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/bcftools.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/bed.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/cellranger.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/cellranger_pipeline.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/cnv.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/cnvkit.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/cnvkit_pipeline.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/gene.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/gsea.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/misc.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/rnaseq.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/stats.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/tcgamaf.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/vcf.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/ns/web.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/bam/CNAClinic.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/bam/CNVpytor.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/bam/ControlFREEC.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cellranger/CellRangerCount.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cellranger/CellRangerSummary.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cellranger/CellRangerVdj.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnv/AneuploidyScore.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnv/AneuploidyScoreSummary.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnv/TMADScoreSummary.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnvkit/CNVkitDiagram.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnvkit/CNVkitHeatmap.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/cnvkit/CNVkitScatter.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/delim/SampleInfo.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/gsea/FGSEA.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/gsea/GSEA.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/CellsDistribution.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/DimPlots.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/MarkersFinder.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/MetaMarkers.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/RadarPlots.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/ScFGSEA.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/SeuratClusterStats.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/SeuratMap2Ref.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/SeuratPreparing.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna/TopExpressingGenes.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna_metabolic_landscape/MetabolicFeatures.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna_metabolic_landscape/MetabolicFeaturesIntraSubset.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayActivity.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/scrna_metabolic_landscape/MetabolicPathwayHeterogeneity.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/CDR3AAPhyschem.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/CloneResidency.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/Immunarch.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/SampleDiversity.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/TCRClusterStats.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/TESSA.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/tcr/VJUsage.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/utils/gsea.liq +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/utils/misc.liq +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/vcf/TruvariBenchSummary.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/reports/vcf/TruvariConsistency.svelte +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bam/BamMerge.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bam/BamSplitChroms.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bam/CNAClinic.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bam/CNVpytor.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bam/ControlFREEC.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bcftools/BcftoolsAnnotate.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bcftools/BcftoolsFilter.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bcftools/BcftoolsSort.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bed/Bed2Vcf.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bed/BedConsensus.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bed/BedLiftOver.sh +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/bed/BedtoolsMerge.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cellranger/CellRangerCount.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cellranger/CellRangerSummary.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cellranger/CellRangerVdj.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnv/AneuploidyScore.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnv/AneuploidyScoreSummary.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnv/TMADScore.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnv/TMADScoreSummary.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitAccess.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitAutobin.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitBatch.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitCall.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitCoverage.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitDiagram.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitFix.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitGuessBaits.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitHeatmap.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitReference.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitScatter.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/CNVkitSegment.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/cnvkit/guess_baits.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/delim/RowsBinder.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/gene/GeneNameConversion.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/gsea/Enrichr.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/gsea/FGSEA.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/gsea/GSEA.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/gsea/PreRank.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/misc/Config2File.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/misc/Str2File.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/plot/Heatmap.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/plot/VennDiagram.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/rnaseq/Simulation-ESCO.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/rnaseq/Simulation-RUVcorr.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/rnaseq/Simulation.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/rnaseq/UnitConversion.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/AnnData2Seurat.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation-celltypist.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation-direct.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation-hitype.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation-sccatch.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation-sctype.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellTypeAnnotation.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/CellsDistribution.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/DimPlots.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ExprImpution-alra.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ExprImpution-rmagic.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ExprImpution-scimpute.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ExprImpution.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/ModuleScoreCalculator.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/RadarPlots.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SCImpute.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/Seurat2AnnData.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats-dimplots.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats-features.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats-hists.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats-ngenes.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats-stats.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClusterStats.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratClustering.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratFilter.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratLoading.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratMap2Ref.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratMetadataMutater.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratSplit.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratSubClustering.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratSubset.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/SeuratTo10X.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/TopExpressingGenes.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/Write10X.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/celltypist-wrapper.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna/sctype.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna_metabolic_landscape/MetabolicFeatures.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna_metabolic_landscape/MetabolicFeaturesIntraSubset.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna_metabolic_landscape/MetabolicPathwayActivity.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/scrna_metabolic_landscape/MetabolicPathwayHeterogeneity.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/snp/PlinkSimulation.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/stats/ChowTest.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/stats/DiffCoexpr.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/stats/LiquidAssoc.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/stats/MetaPvalue.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcgamaf/Maf2Vcf.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcgamaf/MafAddChr.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcgamaf/maf2vcf.pl +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Attach2Seurat.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/CDR3AAPhyschem.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/CloneResidency.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/CloneSizeQQPlot.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/GIANA/GIANA.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/GIANA/GIANA4.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/GIANA/Imgt_Human_TRBV.fasta +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/GIANA/query.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-basic.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-clonality.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-diversity.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-geneusage.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-kmer.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-overlap.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-spectratyping.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-tracking.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch-vjjunc.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/Immunarch2VDJtools.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/ImmunarchFilter.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/ImmunarchLoading.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/ImmunarchSplitIdents.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/SampleDiversity.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TCRClusterStats.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TCRClustering.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TCRDock.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/Atchley_factors.csv +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/BriseisEncoder.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/MCMC_control.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/TrainedEncoder.h5 +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/fixed_b.csv +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/initialization.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/post_analysis.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/real_data.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/update.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/TESSA_source/utility.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/VJUsage.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/immunarch-patched.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/tcr/vdjtools-patch.sh +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/TruvariBench.sh +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/TruvariBenchSummary.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/TruvariConsistency.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/Vcf2Bed.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfAnno.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfDownSample.sh +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfFilter.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfFix.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfFix_utils.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfIndex.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfIntersect.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfLiftOver.sh +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/vcf/VcfSplitSamples.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/web/Download.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/scripts/web/DownloadList.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/__init__.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/caching.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/common_docstrs.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/gene.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/gene.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/gsea.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/io.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/misc.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/misc.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/mutate_helpers.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/plot.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/reference.py +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/rnaseq.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/single_cell.R +0 -0
- {biopipen-0.27.2 → biopipen-0.27.4}/biopipen/utils/vcf.py +0 -0
|
@@ -1,6 +1,6 @@
|
|
|
1
1
|
Metadata-Version: 2.1
|
|
2
2
|
Name: biopipen
|
|
3
|
-
Version: 0.27.
|
|
3
|
+
Version: 0.27.4
|
|
4
4
|
Summary: Bioinformatics processes/pipelines that can be run from `pipen run`
|
|
5
5
|
License: MIT
|
|
6
6
|
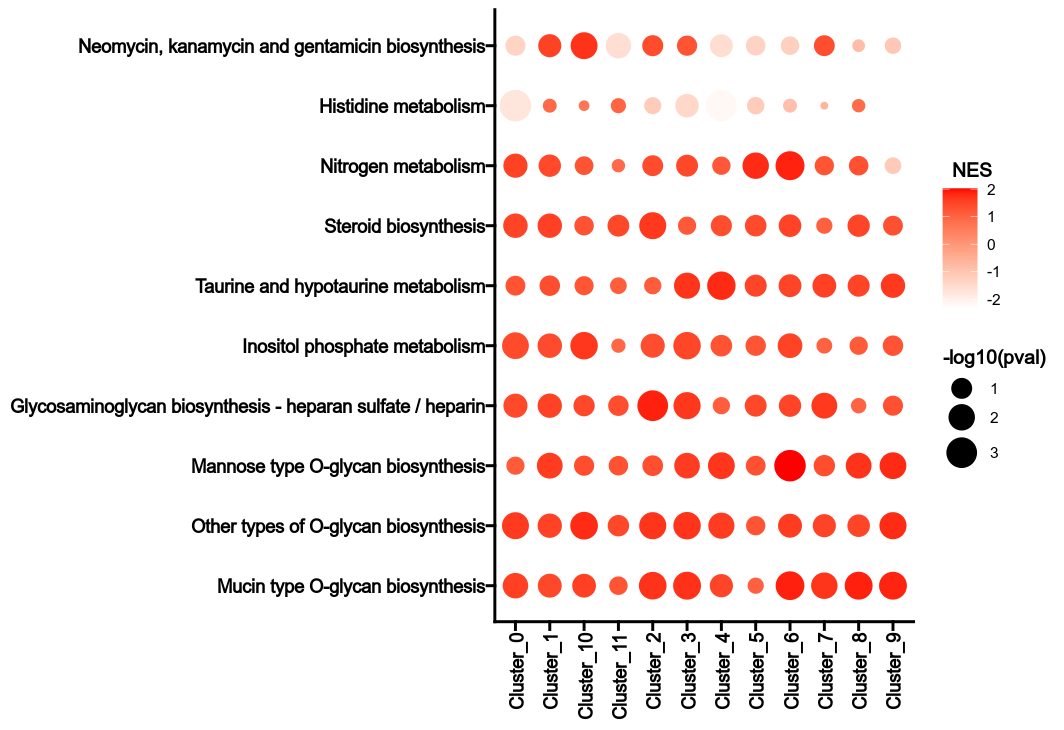
Author: pwwang
|
|
@@ -17,6 +17,6 @@ Requires-Dist: datar[pandas] (>=0.15.6,<0.16.0)
|
|
|
17
17
|
Requires-Dist: pipen-board[report] (>=0.15,<0.16)
|
|
18
18
|
Requires-Dist: pipen-cli-run (>=0.13,<0.14)
|
|
19
19
|
Requires-Dist: pipen-filters (>=0.12,<0.13)
|
|
20
|
-
Requires-Dist: pipen-poplog (>=0.1,<0.2)
|
|
20
|
+
Requires-Dist: pipen-poplog (>=0.1.2,<0.2.0)
|
|
21
21
|
Requires-Dist: pipen-runinfo (>=0.6,<0.7) ; extra == "runinfo"
|
|
22
22
|
Requires-Dist: pipen-verbose (>=0.11,<0.12)
|
|
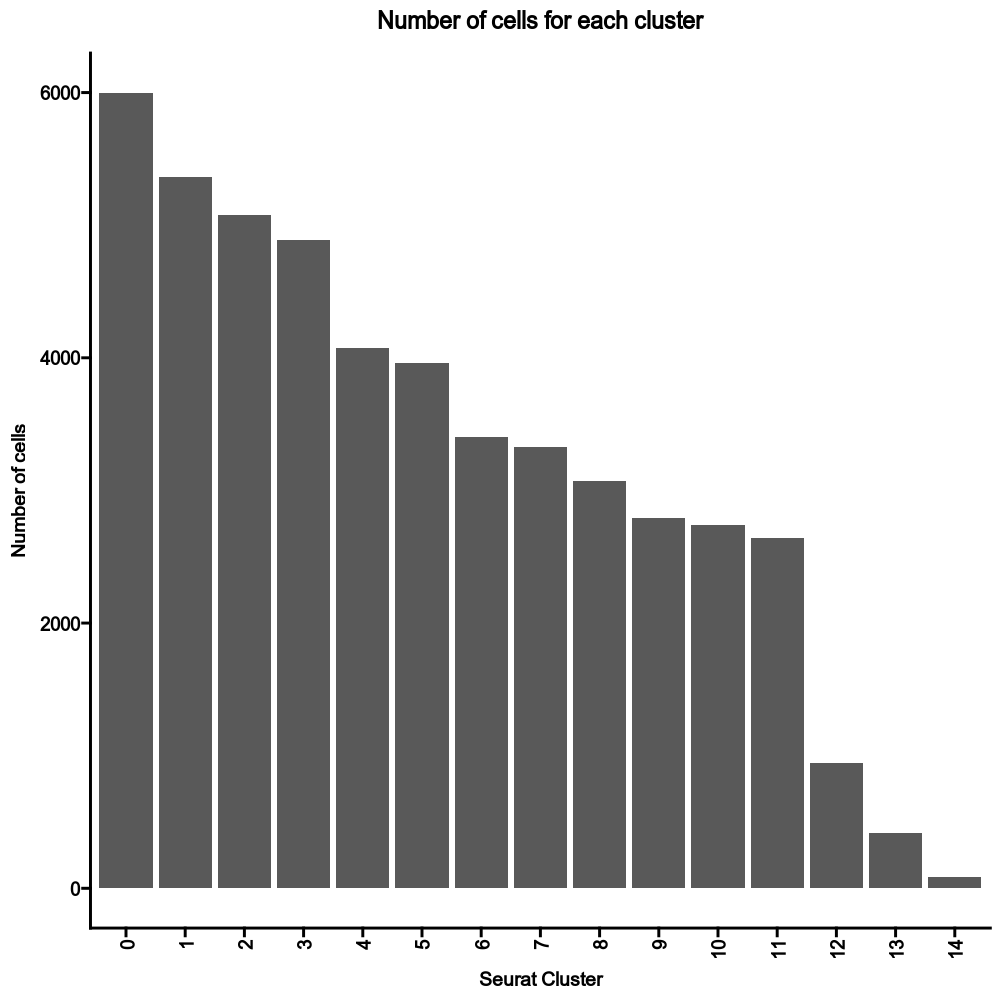
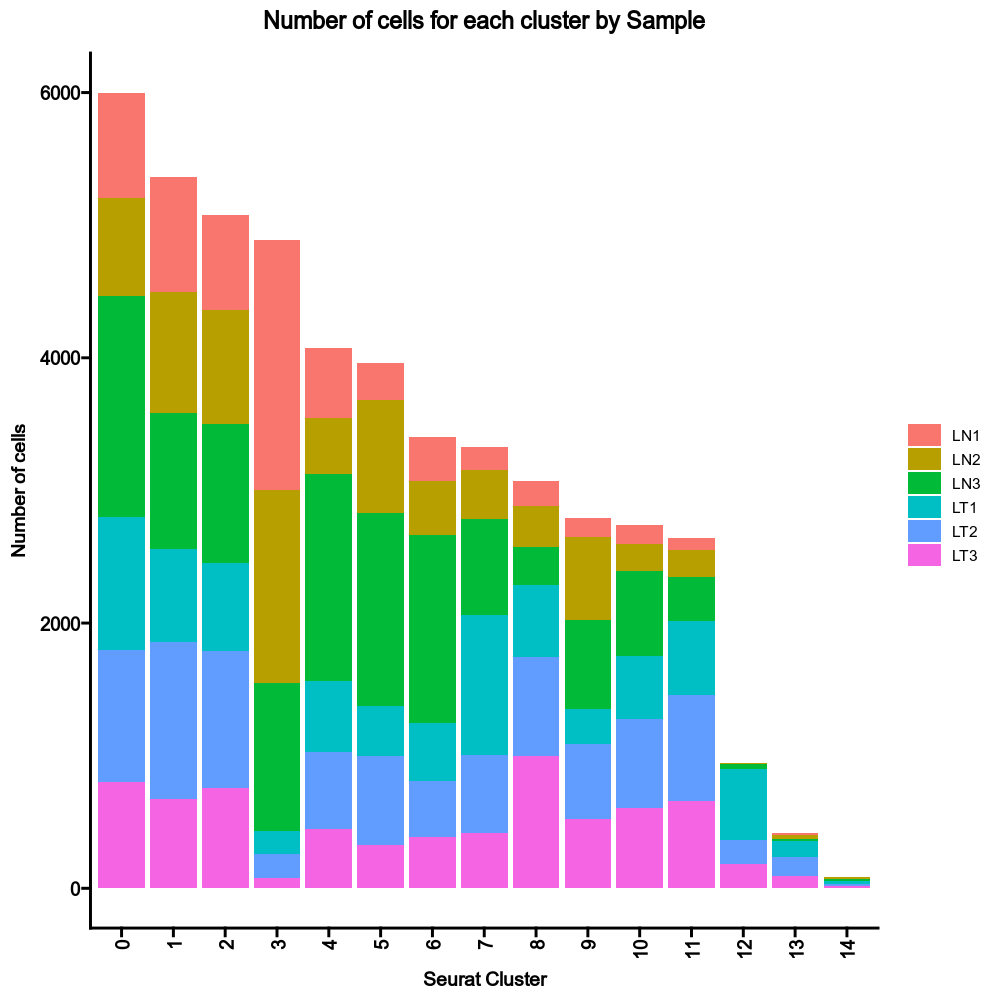
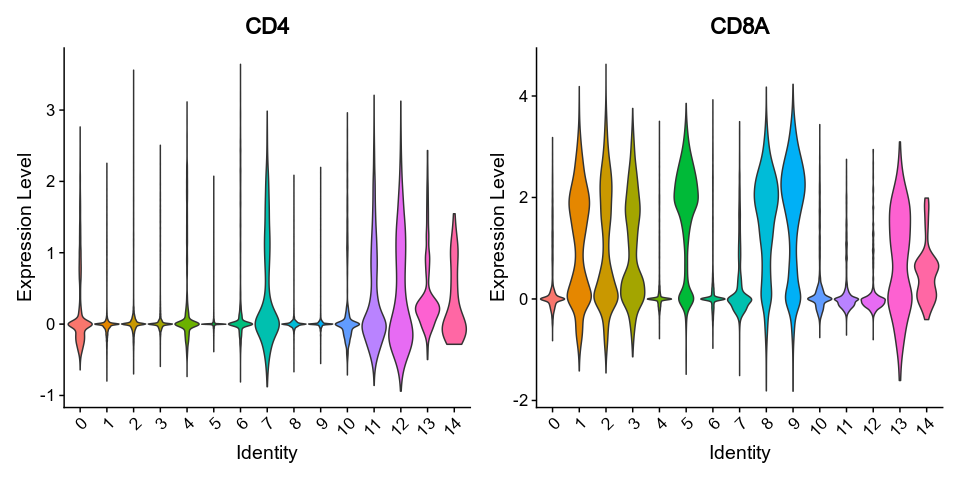
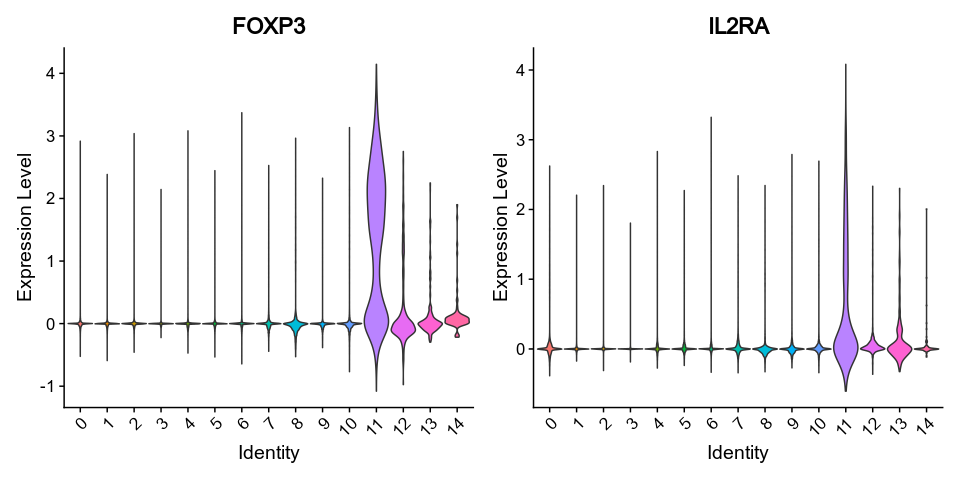
@@ -0,0 +1 @@
|
|
|
1
|
+
__version__ = "0.27.4"
|
|
@@ -114,3 +114,39 @@ class Heatmap(Proc):
|
|
|
114
114
|
"globals": "",
|
|
115
115
|
}
|
|
116
116
|
script = "file://../scripts/plot/Heatmap.R"
|
|
117
|
+
|
|
118
|
+
|
|
119
|
+
class ROC(Proc):
|
|
120
|
+
"""Plot ROC curve using [`plotROC`](https://cran.r-project.org/web/packages/plotROC/vignettes/examples.html).
|
|
121
|
+
|
|
122
|
+
Input:
|
|
123
|
+
infile: The input file for data, tab-separated.
|
|
124
|
+
The first column should be ids of the records (this is optional if `envs.noids` is True).
|
|
125
|
+
The second column should be the labels of the records (1 for positive, 0 for negative).
|
|
126
|
+
If they are not binary, you can specify the positive label by `envs.pos_label`.
|
|
127
|
+
From the third column, it should be the scores of the different models.
|
|
128
|
+
|
|
129
|
+
Output:
|
|
130
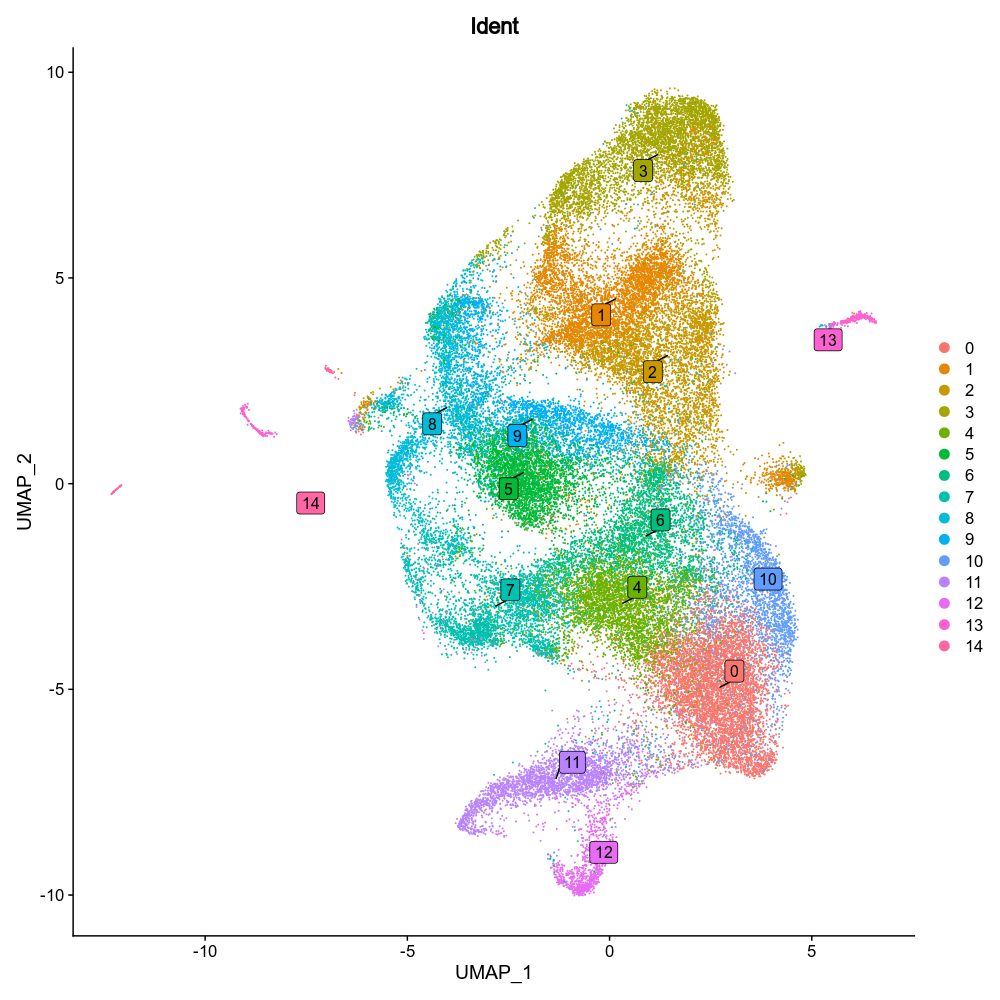
|
+
outfile: The output figure file
|
|
131
|
+
|
|
132
|
+
Envs:
|
|
133
|
+
noids: Whether the input file has ids (first column) or not.
|
|
134
|
+
pos_label: The positive label.
|
|
135
|
+
ci: Whether to use `geom_rocci()` instead of `geom_roc()`.
|
|
136
|
+
devpars: The parameters for `png()`
|
|
137
|
+
args: Additional arguments for `geom_roc()` or `geom_rocci()` if `envs.ci` is True.
|
|
138
|
+
style_roc: Arguments for `style_roc()`
|
|
139
|
+
""" # noqa: E501
|
|
140
|
+
input = "infile:file"
|
|
141
|
+
output = "outfile:file:{{in.infile | stem}}.roc.png"
|
|
142
|
+
lang = config.lang.rscript
|
|
143
|
+
envs = {
|
|
144
|
+
"noids": False,
|
|
145
|
+
"pos_label": 1,
|
|
146
|
+
"ci": False,
|
|
147
|
+
"devpars": {"res": 100, "width": 750, "height": 600},
|
|
148
|
+
"args": {"labels": False},
|
|
149
|
+
"style_roc": {},
|
|
150
|
+
"show_auc": True,
|
|
151
|
+
}
|
|
152
|
+
script = "file://../scripts/plot/ROC.R"
|
|
@@ -201,6 +201,13 @@ class SeuratPreparing(Proc):
|
|
|
201
201
|
- scvi: Same as `scVIIntegration`.
|
|
202
202
|
- <more>: See <https://satijalab.org/seurat/reference/integratelayers>
|
|
203
203
|
|
|
204
|
+
DoubletFinder (ns): Arguments to run [`DoubletFinder`](https://github.com/chris-mcginnis-ucsf/DoubletFinder).
|
|
205
|
+
See also <https://demultiplexing-doublet-detecting-docs.readthedocs.io/en/latest/DoubletFinder.html>.
|
|
206
|
+
To disable `DoubletFinder`, set `envs.DoubletFinder` to `None` or `False`; or set `pcs` to `0`.
|
|
207
|
+
- PCs (type=int): Number of PCs to use for 'doubletFinder' function.
|
|
208
|
+
- doublets (type=float): Number of expected doublets as a proportion of the pool size.
|
|
209
|
+
- pN (type=float): Number of doublets to simulate as a proportion of the pool size.
|
|
210
|
+
|
|
204
211
|
Requires:
|
|
205
212
|
r-seurat:
|
|
206
213
|
- check: {{proc.lang}} <(echo "library(Seurat)")
|
|
@@ -227,6 +234,7 @@ class SeuratPreparing(Proc):
|
|
|
227
234
|
"min_cells": 5,
|
|
228
235
|
},
|
|
229
236
|
"IntegrateLayers": {"method": "harmony"},
|
|
237
|
+
"DoubletFinder": {"PCs": 0, "pN": 0.25, "doublets": 0.075},
|
|
230
238
|
}
|
|
231
239
|
script = "file://../scripts/scrna/SeuratPreparing.R"
|
|
232
240
|
plugin_opts = {
|
|
@@ -405,7 +413,7 @@ class SeuratClusterStats(Proc):
|
|
|
405
413
|
nCells_All = { }
|
|
406
414
|
```
|
|
407
415
|
|
|
408
|
-
{: width="80%" }
|
|
416
|
+
{: width="80%" }
|
|
409
417
|
|
|
410
418
|
### Number of cells in each cluster by groups
|
|
411
419
|
|
|
@@ -414,7 +422,7 @@ class SeuratClusterStats(Proc):
|
|
|
414
422
|
nCells_Sample = { group-by = "Sample" }
|
|
415
423
|
```
|
|
416
424
|
|
|
417
|
-
{: width="80%" }
|
|
425
|
+
{: width="80%" }
|
|
418
426
|
|
|
419
427
|
### Violin plots for the gene expressions
|
|
420
428
|
|
|
@@ -427,8 +435,8 @@ class SeuratClusterStats(Proc):
|
|
|
427
435
|
vlnplots_1 = { features = ["FOXP3", "IL2RA"], pt-size = 0, kind = "vln" }
|
|
428
436
|
```
|
|
429
437
|
|
|
430
|
-
{: width="80%" }
|
|
431
|
-
{: width="80%" }
|
|
438
|
+
{: width="80%" }
|
|
439
|
+
{: width="80%" }
|
|
432
440
|
|
|
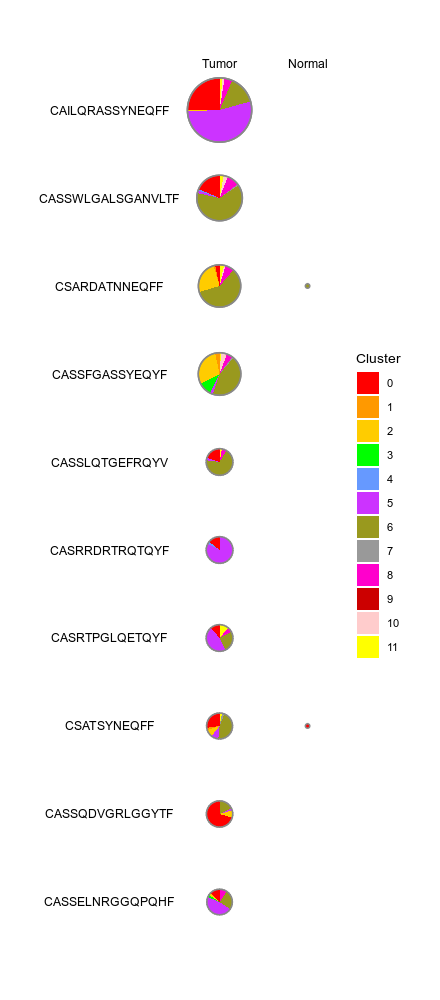
433
441
|
### Dimension reduction plot with labels
|
|
434
442
|
|
|
@@ -439,7 +447,7 @@ class SeuratClusterStats(Proc):
|
|
|
439
447
|
repel = true
|
|
440
448
|
```
|
|
441
449
|
|
|
442
|
-
{: width="80%" }
|
|
450
|
+
{: width="80%" }
|
|
443
451
|
|
|
444
452
|
Input:
|
|
445
453
|
srtobj: The seurat object loaded by `SeuratClustering`
|
|
@@ -849,7 +857,7 @@ class CellsDistribution(Proc):
|
|
|
849
857
|
group_order = [ "Tumor", "Normal" ]
|
|
850
858
|
```
|
|
851
859
|
|
|
852
|
-

|
|
860
|
+
|
|
853
861
|
|
|
854
862
|
Input:
|
|
855
863
|
srtobj: The seurat object in RDS format
|
|
@@ -1862,7 +1870,7 @@ class RadarPlots(Proc):
|
|
|
1862
1870
|
|
|
1863
1871
|
Then we will have a radar plots like this:
|
|
1864
1872
|
|
|
1865
|
-

|
|
1873
|
+
|
|
1866
1874
|
|
|
1867
1875
|
We can use `each` to separate the cells into different cases:
|
|
1868
1876
|
|
|
@@ -1874,7 +1882,7 @@ class RadarPlots(Proc):
|
|
|
1874
1882
|
|
|
1875
1883
|
Then we will have two radar plots, one for `Pre` and one for `Post`:
|
|
1876
1884
|
|
|
1877
|
-

|
|
1885
|
+
|
|
1878
1886
|
|
|
1879
1887
|
Using `cluster_order` to change the order of the clusters and show only the first 3 clusters:
|
|
1880
1888
|
|
|
@@ -1885,7 +1893,7 @@ class RadarPlots(Proc):
|
|
|
1885
1893
|
breaks = [0, 50, 100] # also change the breaks
|
|
1886
1894
|
```
|
|
1887
1895
|
|
|
1888
|
-

|
|
1896
|
+
|
|
1889
1897
|
|
|
1890
1898
|
|
|
1891
1899
|
/// Attention
|
|
@@ -22,11 +22,11 @@ class MetabolicPathwayActivity(Proc):
|
|
|
22
22
|
For each subset, a heatmap and a violin plot will be generated.
|
|
23
23
|
The heatmap shows the pathway activities for each group and each metabolic pathway
|
|
24
24
|
|
|
25
|
-
{: width="80%"}
|
|
25
|
+
{: width="80%"}
|
|
26
26
|
|
|
27
27
|
The violin plot shows the distribution of the pathway activities for each group
|
|
28
28
|
|
|
29
|
-
{: width="45%"}
|
|
29
|
+
{: width="45%"}
|
|
30
30
|
|
|
31
31
|
Envs:
|
|
32
32
|
ntimes (type=int): Number of times to do the permutation
|
|
@@ -294,7 +294,7 @@ class MetabolicPathwayHeterogeneity(Proc):
|
|
|
294
294
|
The heterogeneity can be reflected by the NES values and the p-values in
|
|
295
295
|
different groups for the metabolic pathways.
|
|
296
296
|
|
|
297
|
-

|
|
297
|
+
|
|
298
298
|
|
|
299
299
|
|
|
300
300
|
Envs:
|
|
@@ -71,3 +71,68 @@ class PlinkSimulation(Proc):
|
|
|
71
71
|
"sample_prefix": None,
|
|
72
72
|
}
|
|
73
73
|
script = "file://../scripts/snp/PlinkSimulation.py"
|
|
74
|
+
|
|
75
|
+
|
|
76
|
+
class MatrixEQTL(Proc):
|
|
77
|
+
"""Run Matrix eQTL
|
|
78
|
+
|
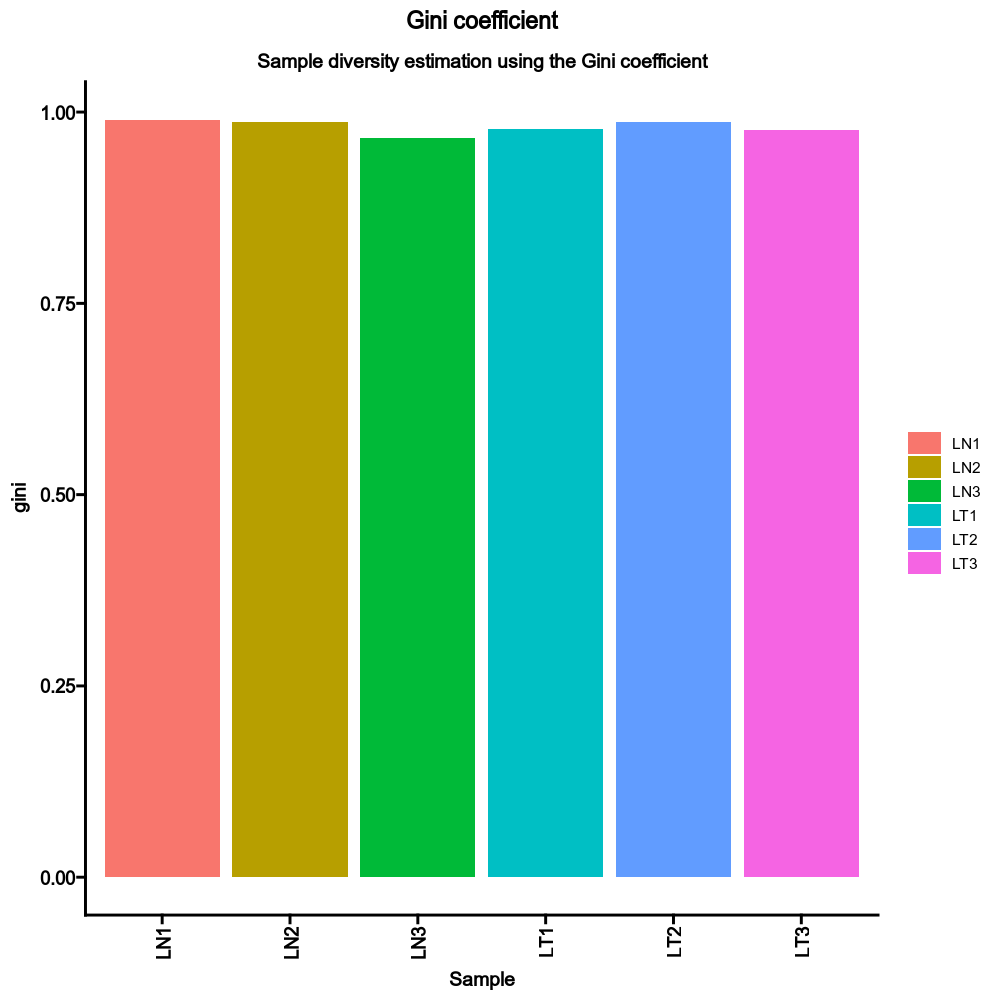
|
79
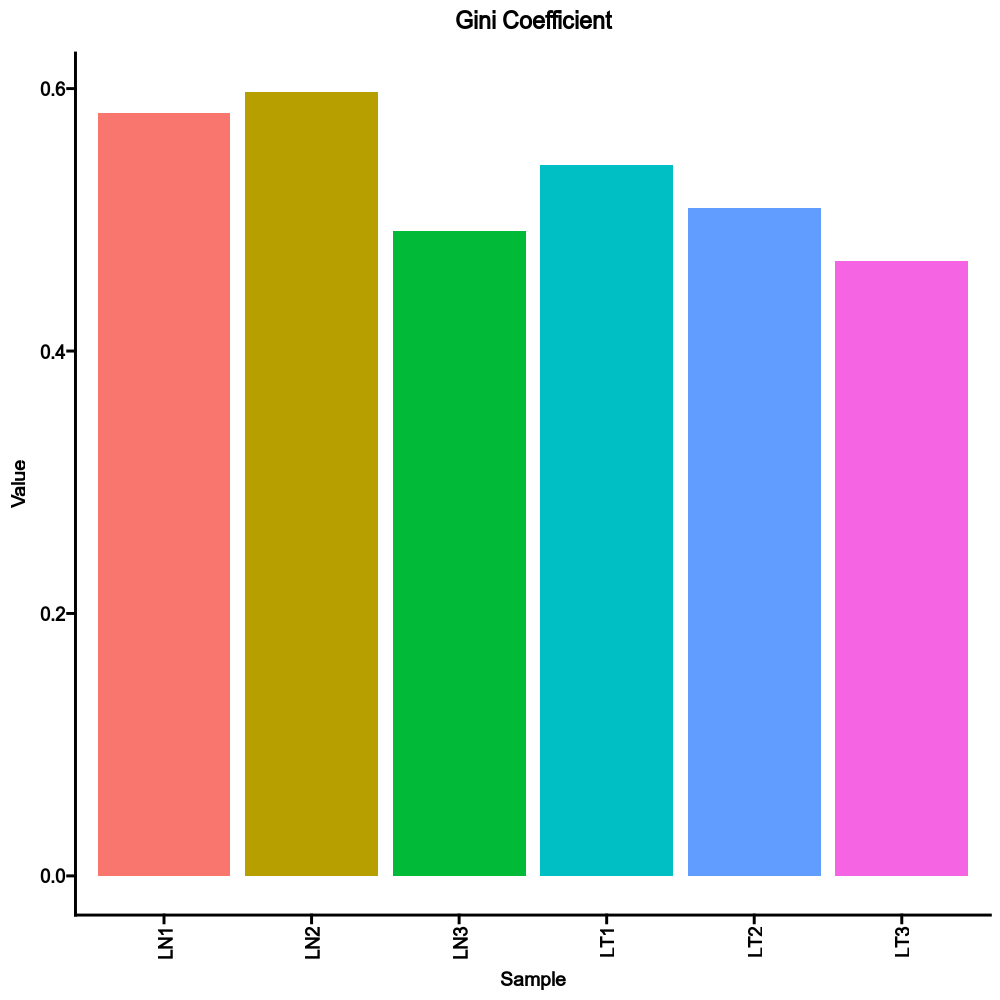
|
+
See also <https://www.bios.unc.edu/research/genomic_software/Matrix_eQTL/>
|
|
80
|
+
|
|
81
|
+
Input:
|
|
82
|
+
geno: Genotype matrix file with rows representing SNPs and columns
|
|
83
|
+
representing samples.
|
|
84
|
+
expr: Expression matrix file with rows representing genes and columns
|
|
85
|
+
representing samples.
|
|
86
|
+
cov: Covariate matrix file with rows representing covariates and columns
|
|
87
|
+
representing samples.
|
|
88
|
+
|
|
89
|
+
Output:
|
|
90
|
+
alleqtls: Matrix eQTL output file
|
|
91
|
+
cisqtls: The cis-eQTL file if `snppos` and `genepos` are provided.
|
|
92
|
+
Otherwise it'll be empty.
|
|
93
|
+
|
|
94
|
+
Envs:
|
|
95
|
+
model (choice): The model to use.
|
|
96
|
+
- `linear`: Linear model
|
|
97
|
+
- `modelLINEAR`: Same as `linear`
|
|
98
|
+
- `anova`: ANOVA model
|
|
99
|
+
- `modelANOVA`: Same as `anova`
|
|
100
|
+
pval (type=float): P-value threshold for eQTLs
|
|
101
|
+
transp (type=float): P-value threshold for trans-eQTLs.
|
|
102
|
+
If cis-eQTLs are not enabled (`snppos` and `genepos` are not set),
|
|
103
|
+
this defaults to 1e-5.
|
|
104
|
+
If cis-eQTLs are enabled, this defaults to `None`, which will disable
|
|
105
|
+
trans-eQTL analysis.
|
|
106
|
+
fdr (flag): Do FDR calculation or not (save memory if not).
|
|
107
|
+
snppos: The path of the SNP position file.
|
|
108
|
+
It could be a BED, GFF, VCF or a tab-delimited file with
|
|
109
|
+
`snp`, `chr`, `pos` as the first 3 columns.
|
|
110
|
+
genepos: The path of the gene position file.
|
|
111
|
+
It could be a BED or GFF file.
|
|
112
|
+
dist (type=int): Distance threshold for cis-eQTLs.
|
|
113
|
+
transpose_geno (flag): If set, the genotype matrix (`in.geno`)
|
|
114
|
+
will be transposed.
|
|
115
|
+
transpose_expr (flag): If set, the expression matrix (`in.expr`)
|
|
116
|
+
will be transposed.
|
|
117
|
+
transpose_cov (flag): If set, the covariate matrix (`in.cov`)
|
|
118
|
+
will be transposed.
|
|
119
|
+
"""
|
|
120
|
+
input = "geno:file, expr:file, cov:file"
|
|
121
|
+
output = [
|
|
122
|
+
"alleqtls:file:{{in.geno | stem}}.alleqtls.txt",
|
|
123
|
+
"cisqtls:file:{{in.geno | stem}}.cisqtls.txt",
|
|
124
|
+
]
|
|
125
|
+
lang = config.lang.rscript
|
|
126
|
+
envs = {
|
|
127
|
+
"model": "linear",
|
|
128
|
+
"pval": 1e-3,
|
|
129
|
+
"transp": None,
|
|
130
|
+
"fdr": False,
|
|
131
|
+
"snppos": None,
|
|
132
|
+
"genepos": config.ref.refgene,
|
|
133
|
+
"dist": 250000,
|
|
134
|
+
"transpose_geno": False,
|
|
135
|
+
"transpose_expr": False,
|
|
136
|
+
"transpose_cov": False,
|
|
137
|
+
}
|
|
138
|
+
script = "file://../scripts/snp/MatrixEQTL.R"
|
|
@@ -923,7 +923,7 @@ class CloneResidency(Proc):
|
|
|
923
923
|
|
|
924
924
|
- Residency plots showing the residency of clones in the two groups
|
|
925
925
|
|
|
926
|
-

|
|
926
|
+
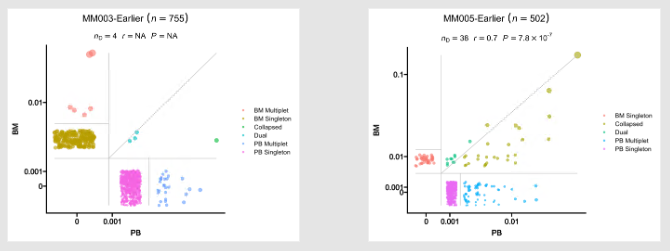
|
|
927
927
|
|
|
928
928
|
The points in the plot are jittered to avoid overplotting. The x-axis is the residency in the first group and
|
|
929
929
|
the y-axis is the residency in the second group. The size of the points are relative to the normalized size of
|
|
@@ -943,7 +943,7 @@ class CloneResidency(Proc):
|
|
|
943
943
|
|
|
944
944
|
- Venn diagrams showing the overlap of the clones in the two groups
|
|
945
945
|
|
|
946
|
-
{: width="60%"}
|
|
946
|
+
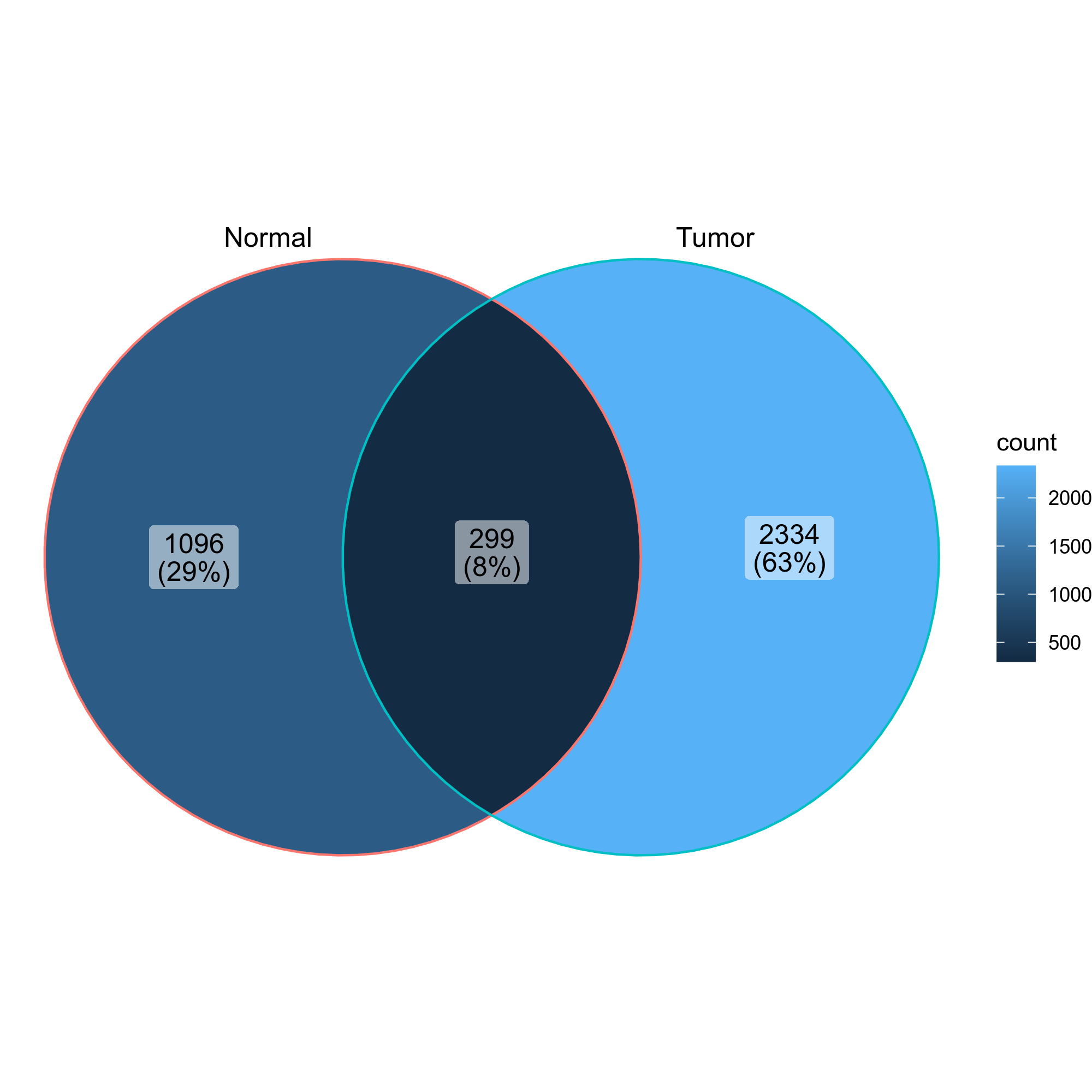{: width="60%"}
|
|
947
947
|
|
|
948
948
|
Input:
|
|
949
949
|
immdata: The data loaded by `immunarch::repLoad()`
|
|
@@ -1259,7 +1259,7 @@ class TCRClusterStats(Proc):
|
|
|
1259
1259
|
by = "Sample"
|
|
1260
1260
|
```
|
|
1261
1261
|
|
|
1262
|
-
{: width="80%"}
|
|
1262
|
+
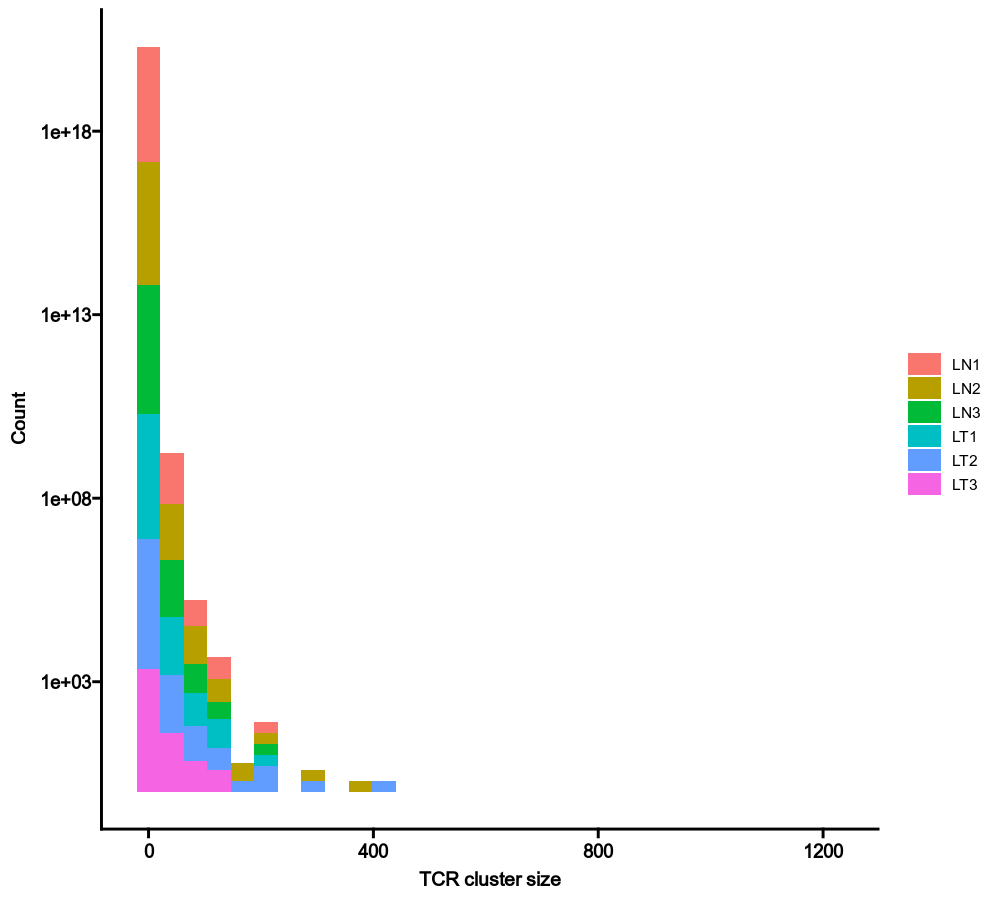{: width="80%"}
|
|
1263
1263
|
|
|
1264
1264
|
### Shared clusters
|
|
1265
1265
|
|
|
@@ -1269,7 +1269,7 @@ class TCRClusterStats(Proc):
|
|
|
1269
1269
|
heatmap_meta = ["region"]
|
|
1270
1270
|
```
|
|
1271
1271
|
|
|
1272
|
-
{: width="80%"}
|
|
1272
|
+
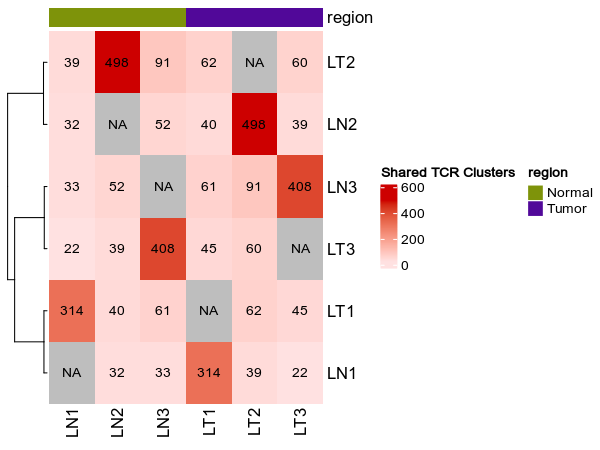{: width="80%"}
|
|
1273
1273
|
|
|
1274
1274
|
### Sample diversity
|
|
1275
1275
|
|
|
@@ -1278,11 +1278,11 @@ class TCRClusterStats(Proc):
|
|
|
1278
1278
|
method = "gini"
|
|
1279
1279
|
```
|
|
1280
1280
|
|
|
1281
|
-
{: width="80%"}
|
|
1281
|
+
{: width="80%"}
|
|
1282
1282
|
|
|
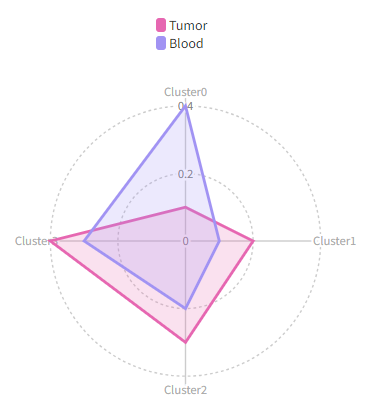
1283
1283
|
Compared to the sample diversity using TCR clones:
|
|
1284
1284
|
|
|
1285
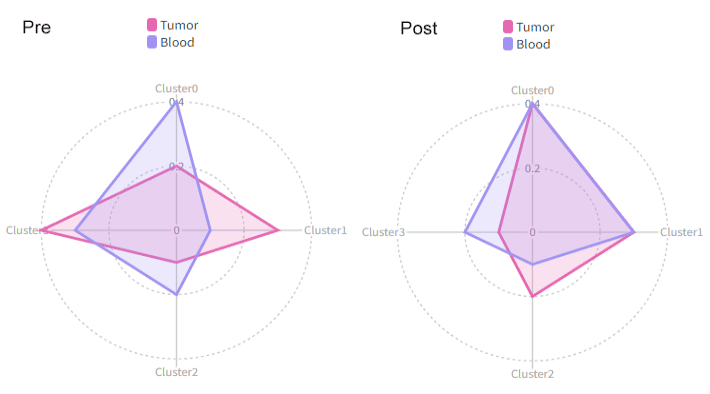
|
-
{: width="80%"}
|
|
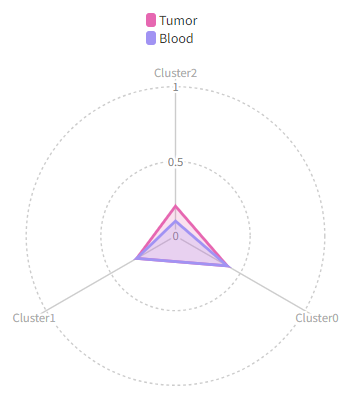
1285
|
+
{: width="80%"}
|
|
1286
1286
|
|
|
1287
1287
|
Input:
|
|
1288
1288
|
immfile: The immunarch object with TCR clusters attached
|
|
@@ -113,14 +113,14 @@ for (name in names(stats)) {
|
|
|
113
113
|
if (stat$plot == "boxplot" || stat$plot == "box") {
|
|
114
114
|
p <- ggplot(data, aes(x=!!group, y=!!sym(stat$on), fill=!!group)) +
|
|
115
115
|
geom_boxplot(position = "dodge") +
|
|
116
|
-
scale_fill_biopipen() +
|
|
116
|
+
scale_fill_biopipen(alpha = .6) +
|
|
117
117
|
xlab("")
|
|
118
118
|
} else if (stat$plot == "violin" ||
|
|
119
119
|
stat$plot == "violinplot" ||
|
|
120
120
|
stat$plot == "vlnplot") {
|
|
121
121
|
p <- ggplot(data, aes(x = !!group, y = !!sym(stat$on), fill=!!group)) +
|
|
122
122
|
geom_violin(position = "dodge") +
|
|
123
|
-
scale_fill_biopipen() +
|
|
123
|
+
scale_fill_biopipen(alpha = .6) +
|
|
124
124
|
xlab("")
|
|
125
125
|
} else if (
|
|
126
126
|
(grepl("violin", stat$plot) || grepl("vln", stat$plot)) &&
|
|
@@ -129,12 +129,12 @@ for (name in names(stats)) {
|
|
|
129
129
|
p <- ggplot(data, aes(x = !!group, y = !!sym(stat$on), fill = !!group)) +
|
|
130
130
|
geom_violin(position = "dodge") +
|
|
131
131
|
geom_boxplot(width = 0.1, position = position_dodge(0.9), fill="white") +
|
|
132
|
-
scale_fill_biopipen() +
|
|
132
|
+
scale_fill_biopipen(alpha = .6) +
|
|
133
133
|
xlab("")
|
|
134
134
|
} else if (stat$plot == "histogram" || stat$plot == "hist") {
|
|
135
135
|
p <- ggplot(data, aes(x = !!sym(stat$on), fill = !!group)) +
|
|
136
136
|
geom_histogram(bins = 10, position = "dodge", alpha = 0.8, color = "white") +
|
|
137
|
-
scale_fill_biopipen()
|
|
137
|
+
scale_fill_biopipen(alpha = .6)
|
|
138
138
|
} else if (stat$plot == "pie" || stat$plot == "piechart") {
|
|
139
139
|
if (is.null(stat$each)) {
|
|
140
140
|
data <- data %>% distinct(!!group, .keep_all = TRUE)
|
|
@@ -157,7 +157,7 @@ for (name in names(stats)) {
|
|
|
157
157
|
fill="#EEEEEE",
|
|
158
158
|
size=4
|
|
159
159
|
) +
|
|
160
|
-
scale_fill_biopipen(name = group) +
|
|
160
|
+
scale_fill_biopipen(alpha = .6, name = group) +
|
|
161
161
|
ggtitle(paste0("# ", stat$on))
|
|
162
162
|
} else if (stat$plot == "bar" || stat$plot == "barplot") {
|
|
163
163
|
if (is.null(stat$each)) {
|
|
@@ -169,7 +169,7 @@ for (name in names(stats)) {
|
|
|
169
169
|
data,
|
|
170
170
|
aes(x = !!group, y = !!sym(count_on), fill = !!group)) +
|
|
171
171
|
geom_bar(stat = "identity") +
|
|
172
|
-
scale_fill_biopipen() +
|
|
172
|
+
scale_fill_biopipen(alpha = .6) +
|
|
173
173
|
ylab(paste0("# ", stat$on))
|
|
174
174
|
} else {
|
|
175
175
|
stop("Unknown plot type: ", stat$plot)
|
|
@@ -0,0 +1,88 @@
|
|
|
1
|
+
|
|
2
|
+
source("{{biopipen_dir}}/utils/misc.R")
|
|
3
|
+
|
|
4
|
+
library(rlang)
|
|
5
|
+
library(ggplot2)
|
|
6
|
+
library(plotROC)
|
|
7
|
+
|
|
8
|
+
infile <- {{in.infile | r}}
|
|
9
|
+
outfile <- {{out.outfile | r}}
|
|
10
|
+
joboutdir <- {{job.outdir | r}}
|
|
11
|
+
noids <- {{envs.noids | r}}
|
|
12
|
+
pos_label <- {{envs.pos_label | r}}
|
|
13
|
+
ci <- {{envs.ci | r}}
|
|
14
|
+
devpars <- {{envs.devpars | r}}
|
|
15
|
+
show_auc <- {{envs.show_auc | r}}
|
|
16
|
+
args <- {{envs.args | r: todot="-"}}
|
|
17
|
+
style_roc_args <- {{envs.style_roc | r: todot="-"}}
|
|
18
|
+
if (!is.null(style_roc_args$theme)) {
|
|
19
|
+
style_roc_args$theme <- eval(parse(text=style_roc_args$theme))
|
|
20
|
+
}
|
|
21
|
+
|
|
22
|
+
data <- read.table(infile, header=TRUE, sep="\t", row.names = NULL, check.names = FALSE, stringsAsFactors=FALSE)
|
|
23
|
+
if (!noids) {
|
|
24
|
+
data <- data[, -1]
|
|
25
|
+
}
|
|
26
|
+
|
|
27
|
+
# Normalize the first column (labels) into 0 and 1.
|
|
28
|
+
# If they are not 0/1, use pos_label to determine the positive class.
|
|
29
|
+
label_col <- colnames(data)[1]
|
|
30
|
+
if (is.character(data[[label_col]])) {
|
|
31
|
+
data[[label_col]] <- as.numeric(data[[label_col]] == pos_label)
|
|
32
|
+
}
|
|
33
|
+
|
|
34
|
+
models <- colnames(data)[2:ncol(data)]
|
|
35
|
+
|
|
36
|
+
if (length(models) > 1) {
|
|
37
|
+
# pivot longer the models, and put the model names into the column 'model'
|
|
38
|
+
data <- melt_roc(data, label_col, colnames(data)[2:ncol(data)])
|
|
39
|
+
} else {
|
|
40
|
+
data <- data.frame(
|
|
41
|
+
D = data[[label_col]],
|
|
42
|
+
M = data[[models]],
|
|
43
|
+
name = rep(models, nrow(data))
|
|
44
|
+
)
|
|
45
|
+
}
|
|
46
|
+
|
|
47
|
+
# Plot the ROC curve
|
|
48
|
+
p <- ggplot(data, aes(d = D, m = M, color = name))
|
|
49
|
+
|
|
50
|
+
if (isTRUE(ci)) {
|
|
51
|
+
p <- p + do.call(geom_rocci, args)
|
|
52
|
+
} else {
|
|
53
|
+
p <- p + do.call(geom_roc, args)
|
|
54
|
+
}
|
|
55
|
+
|
|
56
|
+
p <- p + do.call(style_roc, style_roc_args)
|
|
57
|
+
p <- p + scale_color_biopipen()
|
|
58
|
+
|
|
59
|
+
if (length(models) > 1) {
|
|
60
|
+
p <- p + theme(legend.title = element_blank())
|
|
61
|
+
} else {
|
|
62
|
+
p <- p + theme(legend.position = "none")
|
|
63
|
+
}
|
|
64
|
+
|
|
65
|
+
aucs = calc_auc(p)
|
|
66
|
+
write.table(aucs, file=file.path(joboutdir, "aucs.tsv"), sep="\t", quote=FALSE, row.names=FALSE)
|
|
67
|
+
|
|
68
|
+
if (show_auc) {
|
|
69
|
+
aucs = split(aucs$AUC, aucs$name)
|
|
70
|
+
if (length(aucs) > 1) {
|
|
71
|
+
# Add AUC values to the legend items
|
|
72
|
+
p <- p +
|
|
73
|
+
scale_color_manual(
|
|
74
|
+
values = pal_biopipen()(length(models)),
|
|
75
|
+
labels = sapply(models, function(m) paste(m, " (AUC =", round(aucs[[m]], 2), ")")),
|
|
76
|
+
breaks = models)
|
|
77
|
+
} else {
|
|
78
|
+
p <- p +
|
|
79
|
+
geom_text(
|
|
80
|
+
x = 0.8, y = 0.2, label = paste("AUC =", round(unlist(aucs), 2)),
|
|
81
|
+
color = "black", size = 4)
|
|
82
|
+
}
|
|
83
|
+
}
|
|
84
|
+
|
|
85
|
+
devpars$filename <- outfile
|
|
86
|
+
do.call(png, devpars)
|
|
87
|
+
print(p)
|
|
88
|
+
dev.off()
|
|
@@ -120,7 +120,7 @@ expand_each <- function(name, case) {
|
|
|
120
120
|
pull(case$each) %>% na.omit() %>% unique() %>% as.vector()
|
|
121
121
|
}
|
|
122
122
|
for (each in eachs) {
|
|
123
|
-
by <- make.names(paste0("
|
|
123
|
+
by <- make.names(paste0("..", name, "_", case$each,"_", each))
|
|
124
124
|
srtobj@meta.data <<- srtobj@meta.data %>% mutate(
|
|
125
125
|
!!sym(by) := if_else(
|
|
126
126
|
!!sym(case$each) == each,
|
|
@@ -364,6 +364,16 @@ add_case_report <- function(info, sigmarkers, siggenes) {
|
|
|
364
364
|
}
|
|
365
365
|
}
|
|
366
366
|
|
|
367
|
+
ensure_sobj <- function(expr, allow_empty) {
|
|
368
|
+
tryCatch({ expr }, error = function(e) {
|
|
369
|
+
if (allow_empty) {
|
|
370
|
+
log_warn(" Ignoring this case: {e$message}")
|
|
371
|
+
return(NULL)
|
|
372
|
+
} else {
|
|
373
|
+
stop(e)
|
|
374
|
+
}
|
|
375
|
+
})
|
|
376
|
+
}
|
|
367
377
|
|
|
368
378
|
do_case_findall <- function(casename) {
|
|
369
379
|
# casename
|
|
@@ -382,10 +392,17 @@ do_case_findall <- function(casename) {
|
|
|
382
392
|
# args$min.cells.group <- args$min.cells.group %||% 1
|
|
383
393
|
# args$min.cells.feature <- args$min.cells.feature %||% 1
|
|
384
394
|
# args$min.pct <- args$min.pct %||% 0
|
|
395
|
+
allow_empty = startsWith(case$group.by, "..")
|
|
385
396
|
if (!is.null(case$subset)) {
|
|
386
|
-
args$object <-
|
|
397
|
+
args$object <- ensure_sobj({
|
|
398
|
+
srtobj %>% filter(!!parse_expr(case$subset) & !is.na(!!sym(case$group.by)))
|
|
399
|
+
}, allow_empty)
|
|
400
|
+
if (is.null(args$object)) { return() }
|
|
387
401
|
} else {
|
|
388
|
-
args$object <-
|
|
402
|
+
args$object <- ensure_sobj({
|
|
403
|
+
srtobj %>% filter(!is.na(!!sym(case$group.by)))
|
|
404
|
+
}, allow_empty)
|
|
405
|
+
if (is.null(args$object)) { return() }
|
|
389
406
|
}
|
|
390
407
|
Idents(args$object) <- case$group.by
|
|
391
408
|
|
|
@@ -486,11 +503,19 @@ do_case <- function(casename) {
|
|
|
486
503
|
# sigmarkers
|
|
487
504
|
# rest
|
|
488
505
|
args <- case$rest
|
|
506
|
+
allow_empty = startsWith(case$group.by, "..")
|
|
489
507
|
if (!is.null(case$subset)) {
|
|
490
|
-
args$object <-
|
|
508
|
+
args$object <- ensure_sobj({
|
|
509
|
+
srtobj %>% filter(!!parse_expr(case$subset) & !is.na(!!sym(case$group.by)))
|
|
510
|
+
}, allow_empty)
|
|
511
|
+
if (is.null(args$object)) { return() }
|
|
491
512
|
} else {
|
|
492
|
-
args$object <-
|
|
513
|
+
args$object <- ensure_sobj({
|
|
514
|
+
srtobj %>% filter(!is.na(!!sym(case$group.by)))
|
|
515
|
+
}, allow_empty)
|
|
516
|
+
if (is.null(args$object)) { return() }
|
|
493
517
|
}
|
|
518
|
+
|
|
494
519
|
args$assay <- case$assay
|
|
495
520
|
args$group.by <- case$group.by
|
|
496
521
|
args$ident.1 <- case$ident.1
|
|
@@ -76,7 +76,7 @@ expand_each <- function(name, case) {
|
|
|
76
76
|
pull(case$each) %>% unique() %>% na.omit()
|
|
77
77
|
}
|
|
78
78
|
for (each in eachs) {
|
|
79
|
-
by = make.names(paste0("
|
|
79
|
+
by = make.names(paste0("..", name, "_", case$each, "_", each))
|
|
80
80
|
idents <- case$idents
|
|
81
81
|
if (is.null(idents) || length(idents) == 0) {
|
|
82
82
|
srtobj@meta.data = srtobj@meta.data %>%
|
|
@@ -169,17 +169,31 @@ do_enrich <- function(info, markers, sig) {
|
|
|
169
169
|
}
|
|
170
170
|
}
|
|
171
171
|
|
|
172
|
+
ensure_sobj <- function(expr, allow_empty) {
|
|
173
|
+
tryCatch({ expr }, error = function(e) {
|
|
174
|
+
if (allow_empty) {
|
|
175
|
+
log_warn(" Ignoring this case: {e$message}")
|
|
176
|
+
return(NULL)
|
|
177
|
+
} else {
|
|
178
|
+
stop(e)
|
|
179
|
+
}
|
|
180
|
+
})
|
|
181
|
+
}
|
|
182
|
+
|
|
172
183
|
do_case <- function(casename) {
|
|
173
184
|
log_info("- Dealing with case: {casename} ...")
|
|
174
185
|
info <- casename_info(casename, cases, outdir, create = TRUE)
|
|
175
186
|
case <- cases[[casename]]
|
|
187
|
+
allow_empty = startsWith(case$group_by, "..")
|
|
176
188
|
|
|
177
189
|
if (sum(!is.na(srtobj@meta.data[[case$group_by]])) == 0) {
|
|
178
190
|
msg = "Not enough cells to run tests."
|
|
179
191
|
} else {
|
|
180
|
-
sobj <- srtobj %>% filter(!is.na(!!sym(case$group_by)))
|
|
192
|
+
sobj <- ensure_sobj({ srtobj %>% filter(!is.na(!!sym(case$group_by))) }, allow_empty)
|
|
193
|
+
if (is.null(sobj)) { return() }
|
|
181
194
|
if (!is.null(case$subset)) {
|
|
182
|
-
sobj <-
|
|
195
|
+
sobj <- ensure_sobj({ sobj %>% filter(!!parse_expr(case$subset)) }, allow_empty)
|
|
196
|
+
if (is.null(sobj)) { return() }
|
|
183
197
|
}
|
|
184
198
|
df <- tryCatch({
|
|
185
199
|
GetAssayData(sobj, layer = "data")
|
|
@@ -72,7 +72,7 @@ expand_each <- function(name, case) {
|
|
|
72
72
|
pull(case$each) %>% na.omit() %>% unique() %>% as.vector()
|
|
73
73
|
}
|
|
74
74
|
for (each in eachs) {
|
|
75
|
-
by <- make.names(paste0("
|
|
75
|
+
by <- make.names(paste0("..", name, "_", case$each,"_", each))
|
|
76
76
|
srtobj@meta.data <<- srtobj@meta.data %>%
|
|
77
77
|
mutate(!!sym(by) := if_else(
|
|
78
78
|
!!sym(case$each) == each,
|
|
@@ -97,18 +97,35 @@ log_info("- Expanding cases...")
|
|
|
97
97
|
cases <- expand_cases(cases, defaults, expand_each)
|
|
98
98
|
|
|
99
99
|
|
|
100
|
+
ensure_sobj <- function(expr, allow_empty) {
|
|
101
|
+
tryCatch({ expr }, error = function(e) {
|
|
102
|
+
if (allow_empty) {
|
|
103
|
+
log_warn(" Ignoring this case: {e$message}")
|
|
104
|
+
return(NULL)
|
|
105
|
+
} else {
|
|
106
|
+
stop(e)
|
|
107
|
+
}
|
|
108
|
+
})
|
|
109
|
+
}
|
|
110
|
+
|
|
111
|
+
|
|
100
112
|
do_case <- function(name, case) {
|
|
101
113
|
log_info("- Handling case: {name} ...")
|
|
102
114
|
info <- casename_info(name, cases, outdir, create = TRUE)
|
|
103
115
|
|
|
116
|
+
allow_empty = startsWith(case$group.by, "..")
|
|
104
117
|
# prepare expression matrix
|
|
105
118
|
log_info(" Preparing expression matrix...")
|
|
106
|
-
sobj <- srtobj %>% filter(!is.na(!!sym(case$group.by)))
|
|
119
|
+
sobj <- ensure_sobj({ srtobj %>% filter(!is.na(!!sym(case$group.by))) }, allow_empty)
|
|
120
|
+
if (is.null(sobj)) { return() }
|
|
121
|
+
|
|
107
122
|
if (!is.null(case$subset)) {
|
|
108
|
-
sobj <- sobj %>% filter(!!!parse_exprs(case$subset))
|
|
123
|
+
sobj <- ensure_sobj({ sobj %>% filter(!!!parse_exprs(case$subset)) }, allow_empty)
|
|
124
|
+
if (is.null(sobj)) { return() }
|
|
109
125
|
}
|
|
110
126
|
if (!is.null(case$ident.2)) {
|
|
111
|
-
sobj <- sobj %>% filter(!!sym(case$group.by) %in% c(case$ident.1, case$ident.2))
|
|
127
|
+
sobj <- ensure_sobj({ sobj %>% filter(!!sym(case$group.by) %in% c(case$ident.1, case$ident.2)) }, allow_empty)
|
|
128
|
+
if (is.null(sobj)) { return() }
|
|
112
129
|
}
|
|
113
130
|
|
|
114
131
|
allclasses <- sobj@meta.data[, case$group.by, drop = TRUE]
|